
Richard Meyer
CTO
Summary of Key New Features
VarSome Clinical
- Integration of LOVD component
- CNV VCF sub analysis option for multi-sample analyses
- Integration of PharmCat for pharmacogenomics reporting
- VarSome Picks algorithmic filter for germline and gene list analyses
- Filter analysis results based on custom variant classifications
- Filter variants based on their germline pathogenicity classification points score
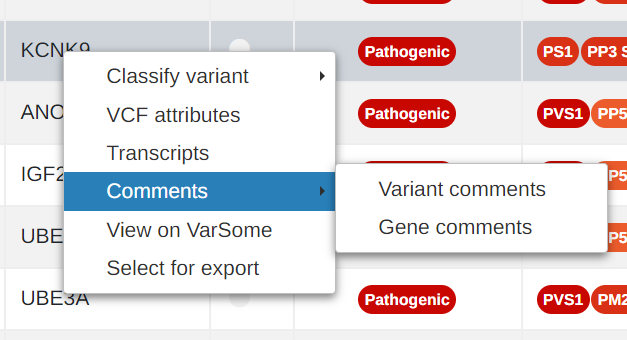
- Add user comments at gene level
VarSome
- CNV Browser improvements
- Region Browser improvements
- Publications improvements
- In-silico predictors improvements
Germline pathogenicity classification
- Modification in the way we filter gnomAD variants
- Integration of LOVD for Clinical and Premium customers
- Further inclusion of ClinGen recommendations for mitochondrial variants
- New in-silico pathogenicity predictor, MaxEntScan, which can classify indels as well as SNVs
Somatic classification
- The somatic classification (based on the AMP guidelines) no longer uses COSMIC when annotating somatic samples
Note: We have removed the 'Old Layout' option and the links back to VarSome from the results table of VarSome Clinical.
New Features
VarSome Clinical
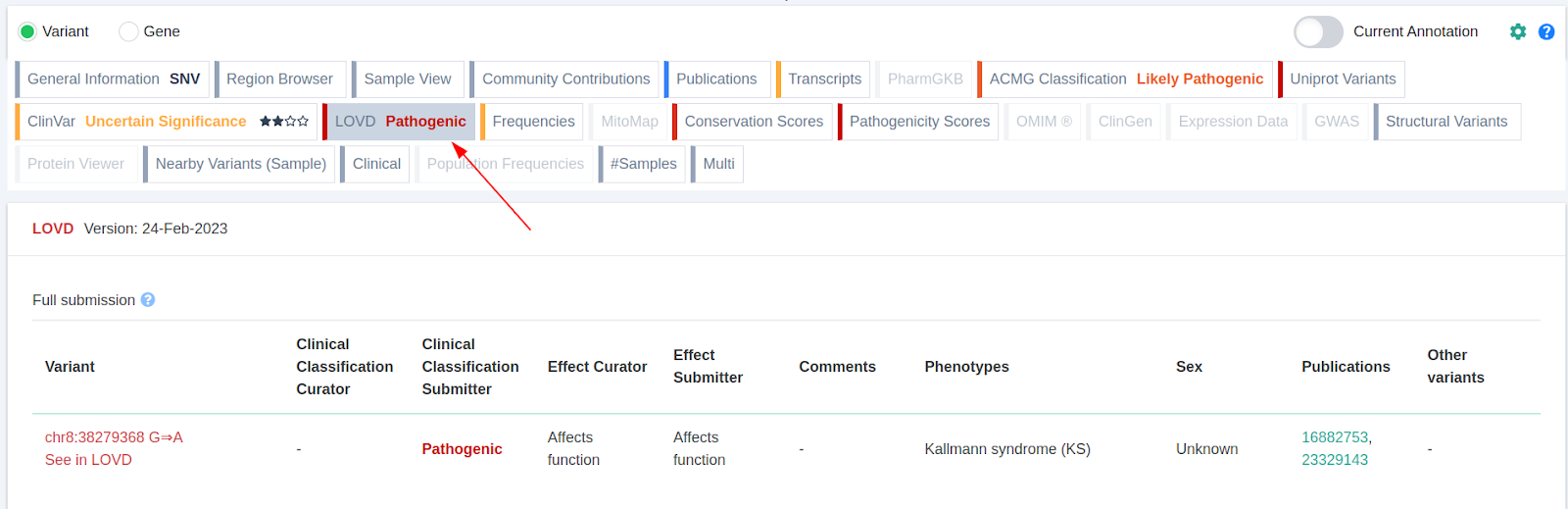
LOVD component
We are pleased to have completed the second phase of the Leiden Open Variation Database (LOVD) integration. In Phase 1 we integrated LOVD data into VarSome Premium and API. For Phase 2, VarSome Clinical users can benefit from these data which have been incorporated into the VarSome’s germline classification as a new source of clinical evidence.
Integration of PharmCat for pharmacogenomics variant annotation Pgx report
PharmCAT is a pharmacogenomics clinical annotation tool that generates a report containing genotype-based prescribing recommendation (PGx report). It has been developed in collaboration with Pharmacogenomics Knowledgebase (PharmGKB) and the former PGRN Statistical Analysis Resource (P-STAR), with input from other groups.
We are now offering the option to automatically generate a PGx report when a WGS sample is run through VarSome Clinical. Supervisors will need to contact our support team and we can then set this up in the group settings at no additional cost.
The PGx report will only be produced when:
- The group setting has been set to PGx report required
- You are running a WGS
- You are running against hg38
For further information please see the PGx help page.
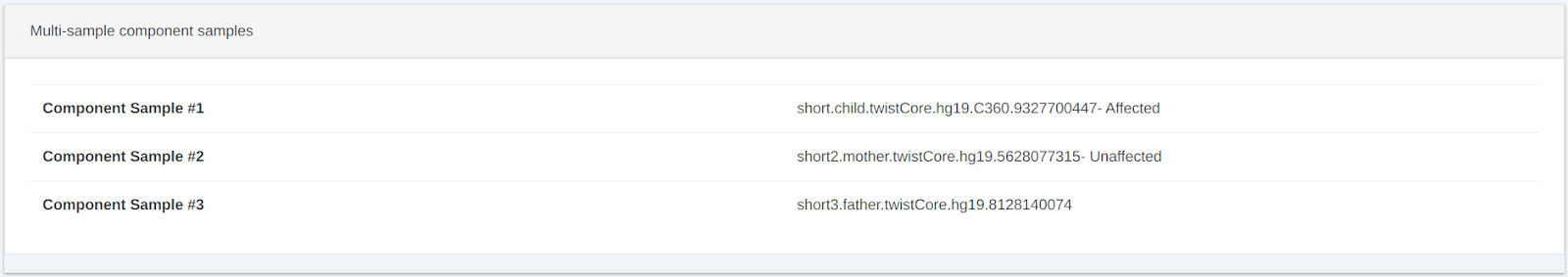
Multi-Sample card added to the card layout to display multi-sample analysis details.
This option was available in the old view of data and has now been included in the card view option.

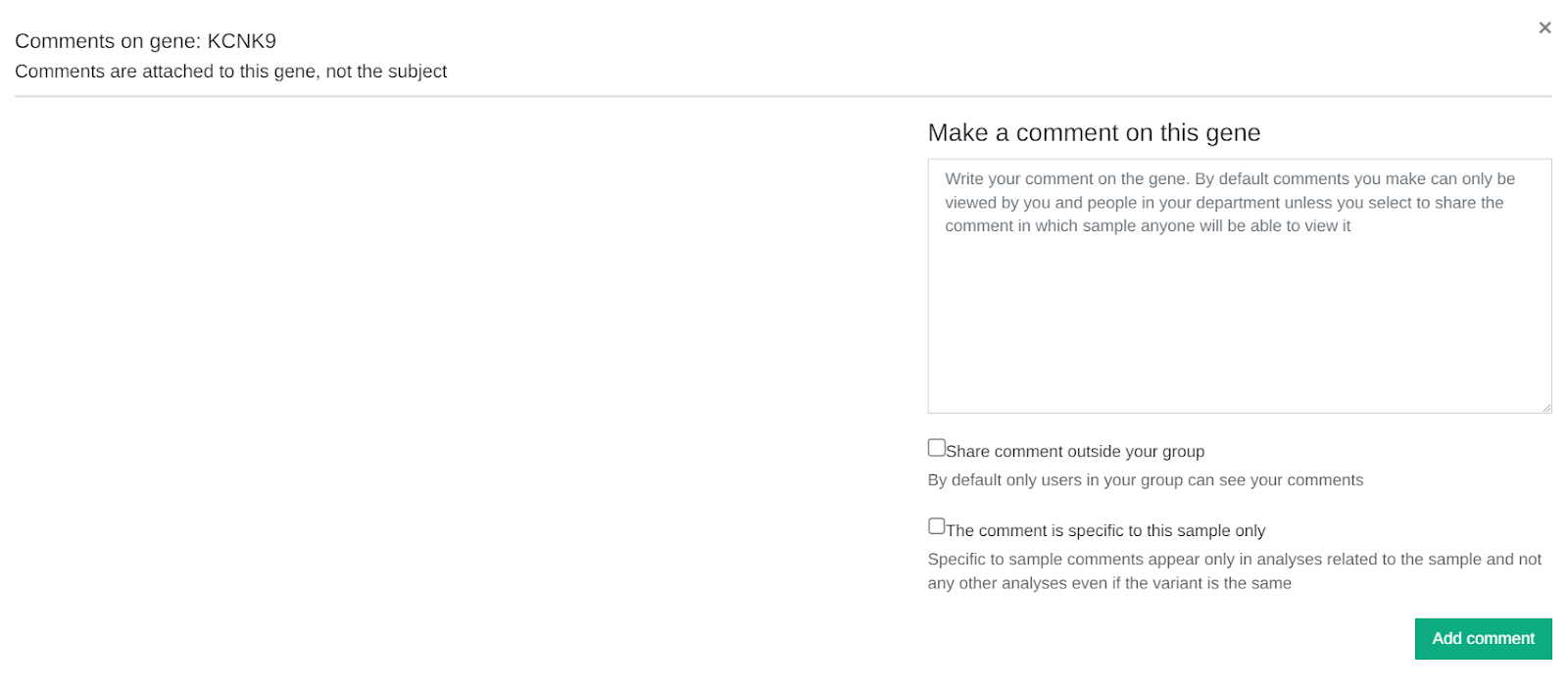
Add user comments at gene level
It is now possible to share comments at gene level in addition to variant level.
Tooltip for allelic balance
A new tooltip has been added to the Allelic Balance and Coverage columns on multi sample and tumor normal analyses that displays which values correspond to which samples.
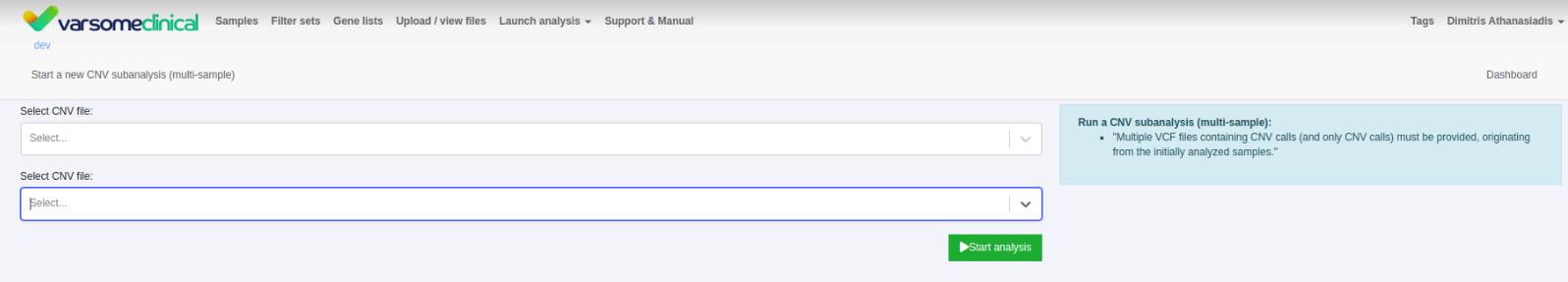
CNV VCF sub analysis option for multiple samples
Previously we could only associate a VCF file with CNVs to be annotated to single samples. The option has now also been added to multi-sample analyses.
Filtering
VarSome Picks - Filtering
VarSome Picks is a new algorithmic filter that takes into account parameters such as phenotype, gene and variant data in order to rank higher the potential causative variants in a disease-specific context.
VarSome Picks will return variants classified as pathogenic, likely pathogenic or VUS found in the top 10 genes* associated to the phenotype(s) selected by the user. The data regarding the gene-disease association and phenotype ontology are coming from the following datasources:
*Note that there is no single source from which all relevant genes can reliably be mined. As a result, extracting data for gene-disease associations from multiple databases is likely to produce a more comprehensive list of variants found to be causative for the disease. Some of those are likely to have been classified as VUS.
VarSome Picks also takes into account:
- Zygosity (variant-related information)
- VarSome germline pathogenicity classification (based on the ACMG criteria)
- Mode of inheritance (gene-related information)
As well as quality parameters of:
- Allelic Balance (threshold for germline variants)
- Coverage in the sample
For further information please review the VarSome Picks help page.
Algorithmic filters now appear in alphabetical order
We have made the launch algorithmic filter page more user friendly, displaying only available filters and in alphabetical order. It is also possible to view the filters that are not applicable for the related samples.
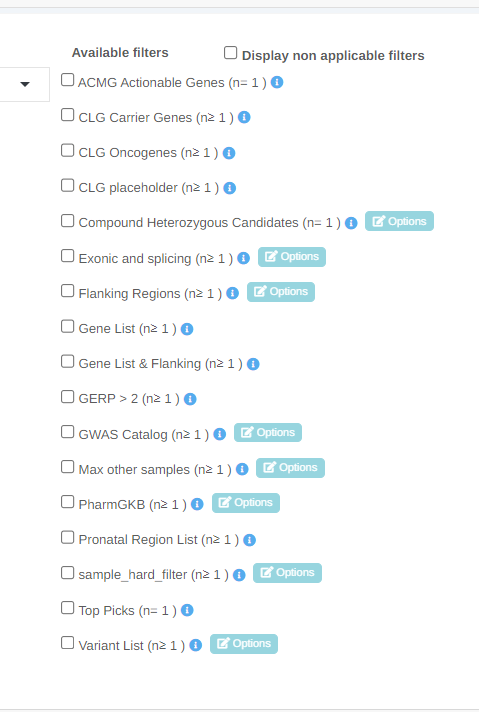
New Filter Options
New - Filter variants by custom classification
New - Germline Classification (ACMG) Points Score filter

Germline classification
We no longer refer to 'ACMG Classification' and instead we use the term 'Germline Classification'
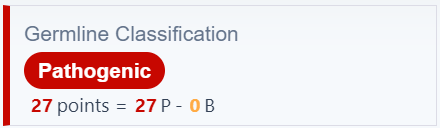
We have further improved our germline pathogenicity classification:
- We have reduced double counting between UniProt and ClinVar where both are mentioned in the same publication.
- Modified filtering of gnomAD variants
We are introducing some changes in the way we use the gnomAD data in our germline variant pathogenicity classification. Our aim is to be as consistent as possible with the way gnomAD data are used.- gnomAD filters: if the variant has an entry in the gnomAD project, we check its associated filter and if it is “PASS”, we will consider the gnomAD frequency of that variant for the VarSome’s Germline Classification.
- Indel coverage: we are using the same approach as used by gnomAD to retrieve the coverage of an indel: the coverage of the first position before the indel of interest.
- Integration of LOVD evidence into the Germline Variant Classification
Clinical evidence is the foundation stone of our germline variant pathogenicity classification. TO date, we have been sourcing this from ClinVar, Uniprot, MitoMap and VarSome user contributions. We are now using LOVD as a new source of clinical evidence. In particular, the new data will be used to trigger the clinical evidence rules (PP5 or BP6) and will be included in the region browser. This is a huge milestone in our that will certainly improve the germline classification. In fact, we have observed that LOVD contains around 150k variants that are not yet reported in ClinVar. - Further inclusion of ClinGen recommendations for mitochondrial variants
In VarSome’s 11.5 release we introduced some improvements into our germline classification for the mitochondrial variants following ClinGen's recommendations. In an effort to keep improving the mitochondrial annotation we are introducing a new rule modification suggested as well by the ClinGen’s mitochondrial expert panel. The rule PM5 will be now used as supporting pathogenic evidence (PM5_Supporting) in non-coding mitochondrial variants where a same nucleotide position as previously established pathogenic variant has been observed. - We have incorporated a new in-silico predictor MaxEntScan for splicing sites for rules BP7 and PP3.
For more information please see: http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html
VarSome
HGVS notation extensions
HGVS notation supported by VarSome has been extended to include single amino acid deletions and duplications within the same exon, which were previously not supported in VarSome queries:
ClinVar Card
We have increased the detail shown on the ClinVar summary card:

Region Browser
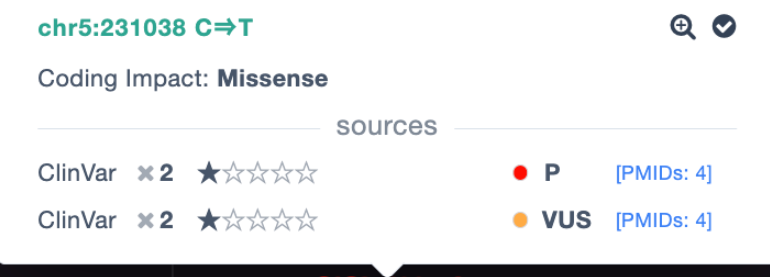
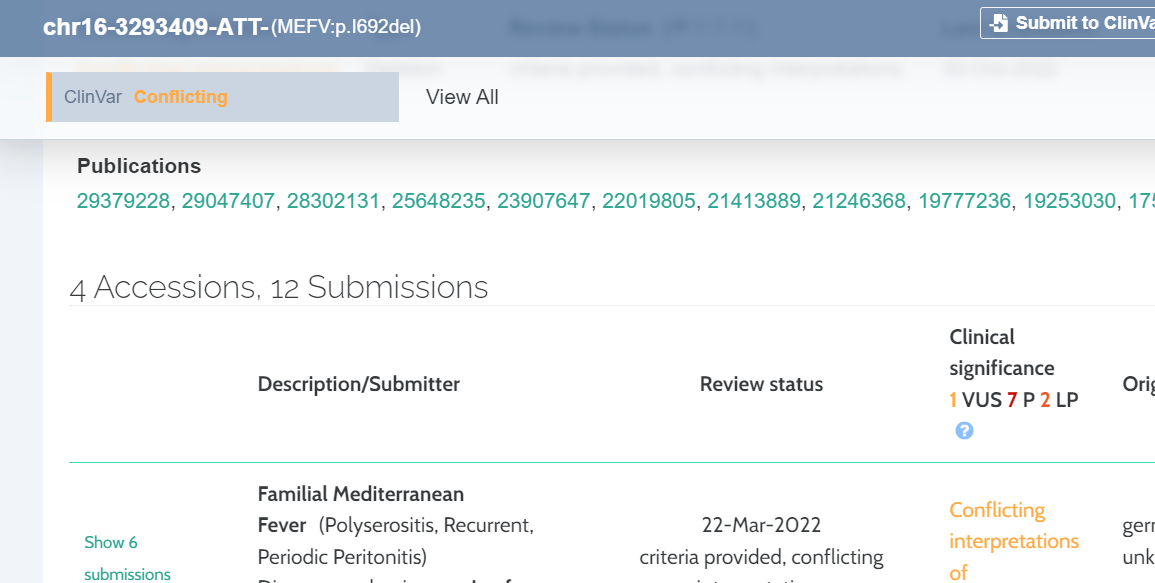
ClinVar conflicting evidence
On the pathogenicity track we have added a tool tip that displays information on variants where there is conflicting ClinVar evidence:
Display User linked publications
We have over 90k variants that have been identified as pathogenic in PubMed articles. We now provide a link to any relevant articles within the Region Browser:
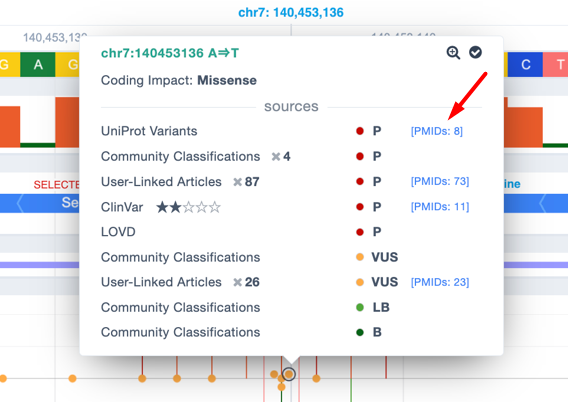
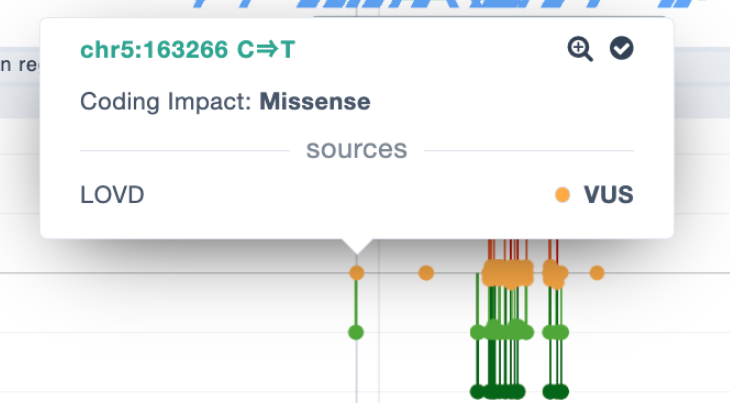
For Premium users, LOVD data have been included in the Region Browser pathogenicity track and included as an option in the filtering:
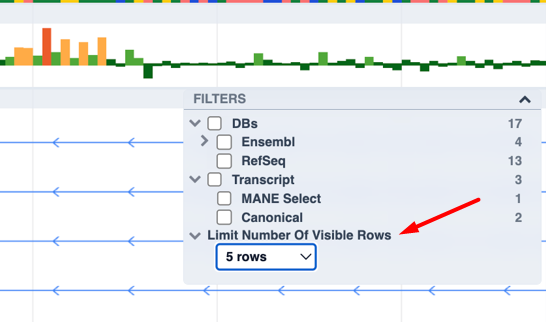
You can now limit the number of visible transcript rows shown:
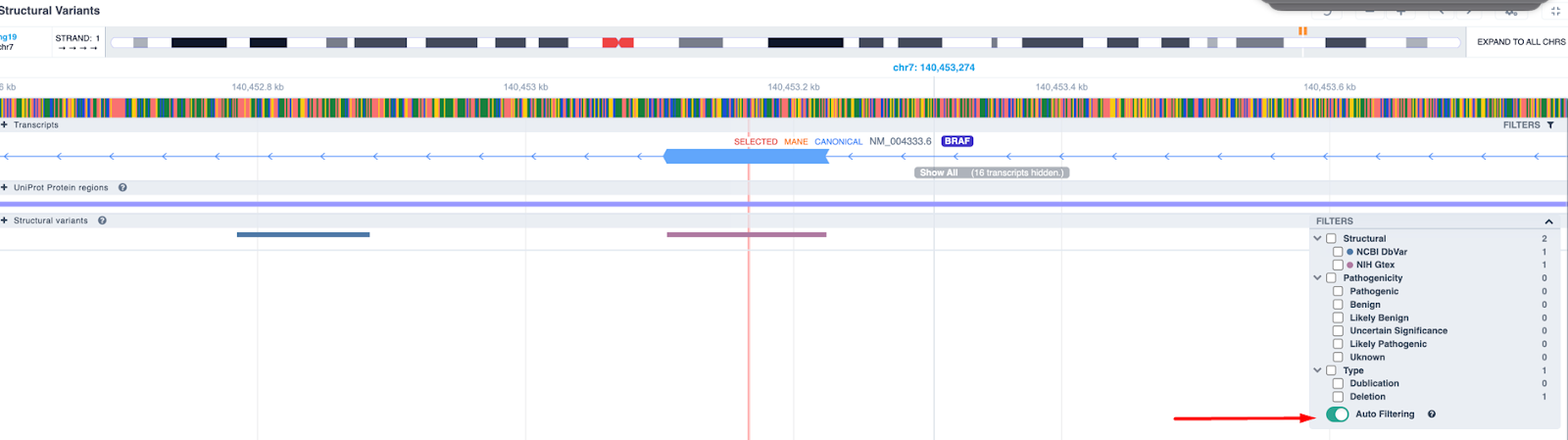
For structural variants, we have added auto filtering according to the visualized region:
CNV Browser
We now display all chromosomes in a single view with the CNVs highlighted:
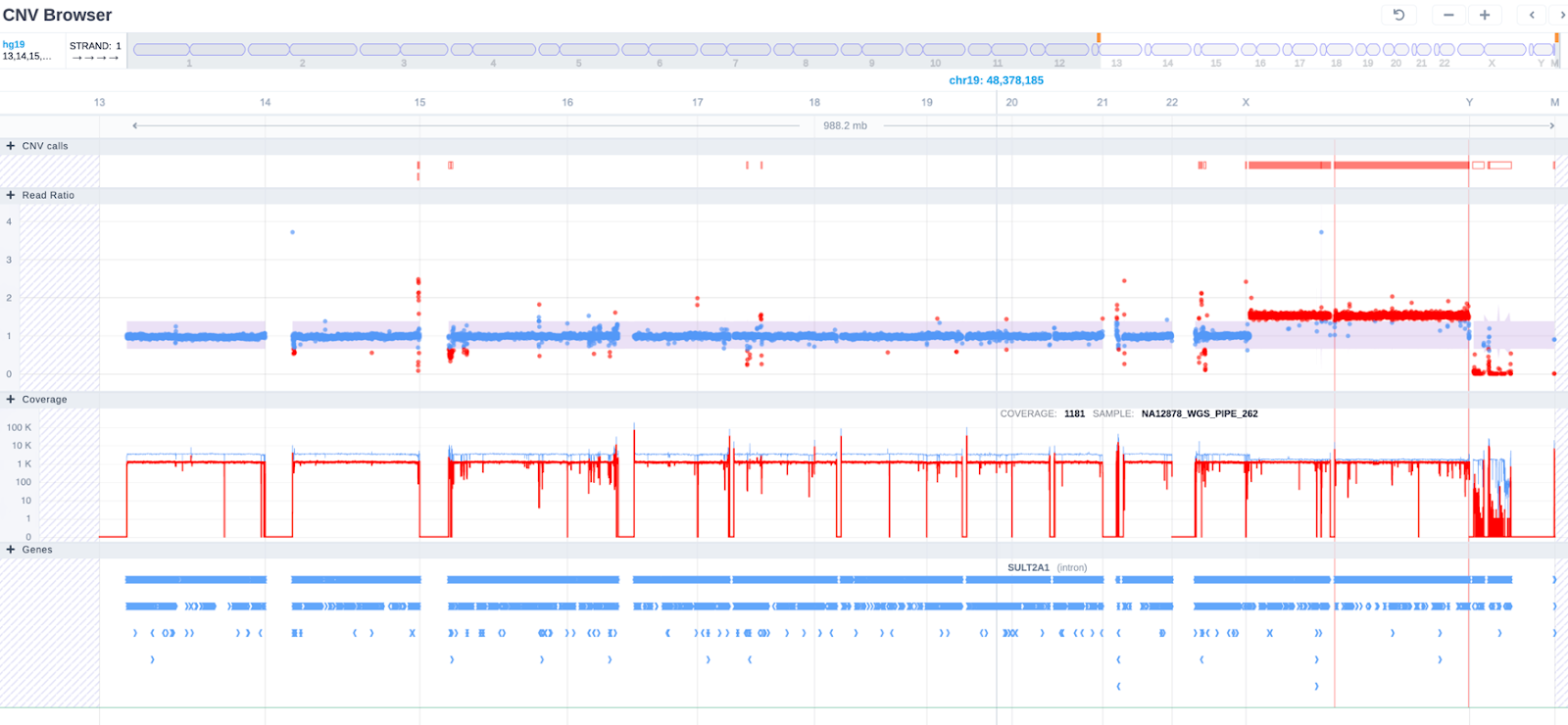
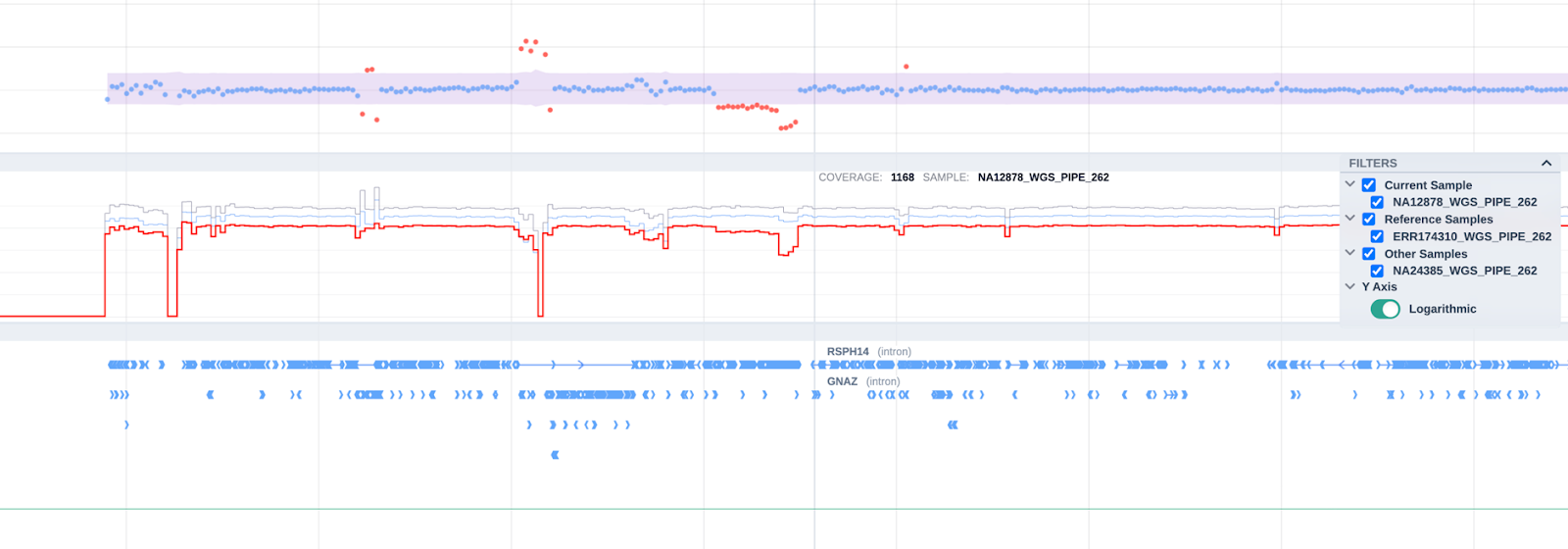
On the coverage track we now highlight the reference sample using color coding and have provided the option to filter by sample:
In-Silico Predictions
Strength of prediction using calibrated thresholds
We have added a new tool-tip to the Score column of the in-silico predictors component (Pathogenicity scores). For a single variant we may have different available transcripts with different predictions and scores. We display all available scores and you can hover over them to see the transcript reference and its calibrated prediction.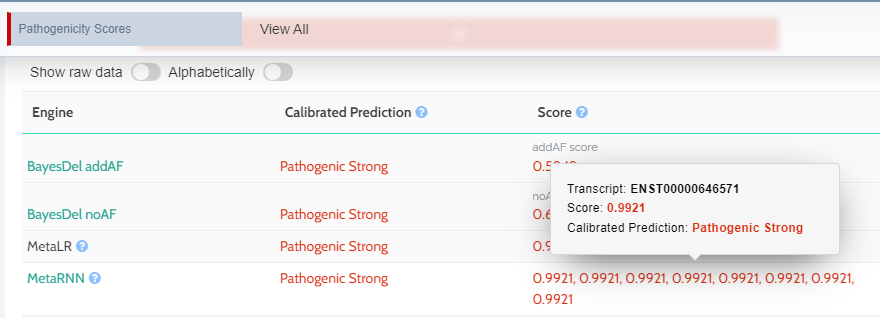
New in-silico pathogenicity predictor for splice sites: Maxentscan
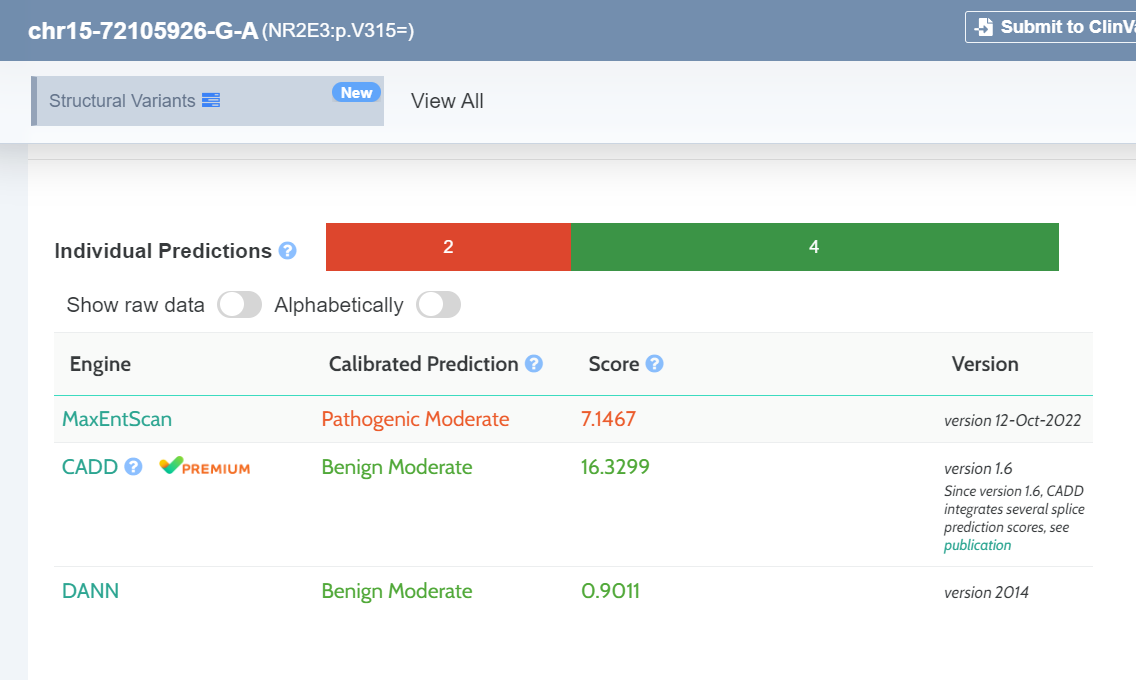
Clinvar - New Clinical Significance summary
A new column has been added to the ClinVar component that displays a color coded summary of the verdicts from different sources.
Publications Improvements
We have improved our publications tagger to better identify cases where keywords can have different meanings in different contexts, plus extended the way we identify and handle acronyms in publications, so that the full word corresponding to an acronym can be identified and highlighted throughout the text.

Support
As ever we hope you find these changes & improvements helpful, we’d love to hear any suggestions you may have, support is available as usual from support@varsome.com.
The VarSome Team.
Submit a Comment