
Richard Meyer
CTO
Key New Features
VarSome Clinical
- OMIM® Integration phenotype matching and OMIM® Inheritance
- On demand annotation of existing sample information
- Improvement to the classification of mitochondrial variants
- Change to variant calling
VarSome
- Remodeled Region Browser
- Additional data extracted for CPIC
- MitoTip and MitImpact pathogenicity scores added to in-silico prediction scores
- Improved ClinVar component
New Features
VarSome Clinical
DNScope and TNhaplotyper2 in joint variant calling
For the sake of consistency with the individual variant calling, we are introducing Sentieon’s DNAscope as the tool of preference for the joint calling of germline, and TNhaplotyper2 in the case of somatic variants. No variants above the noise threshold should be affected.
OMIM® - Phenotype/Inheritance integration
In addition to the recently added OMIM® data source card we have integrated OMIM® data into the clinical platform in the following way:
- Diseases and phenotypes have been merged and we now only refer to phenotypes.
- Filter suggested phenotypes by source when typing a term in the Phenotypes box.
- Select either MONDO, OMIM® or HPO terms or just OMIM® in phenotype matching (displayed in the phenotype column)
- ACMG classification can now use OMIM®, or all data sources, for mode of inheritance of a gene.
For further information please read the OMIM® integration help document.
Live On Demand Annotation of Existing Germline Clinical Samples
It is now possible, for germline samples, to obtain the full, current data that VarSome Premium users would see for all card data sources, rather than just seeing the data stored at the time of the Sample Run.

To avoid confusion The table heading will make it very clear when you are using the 'Current' annotation.
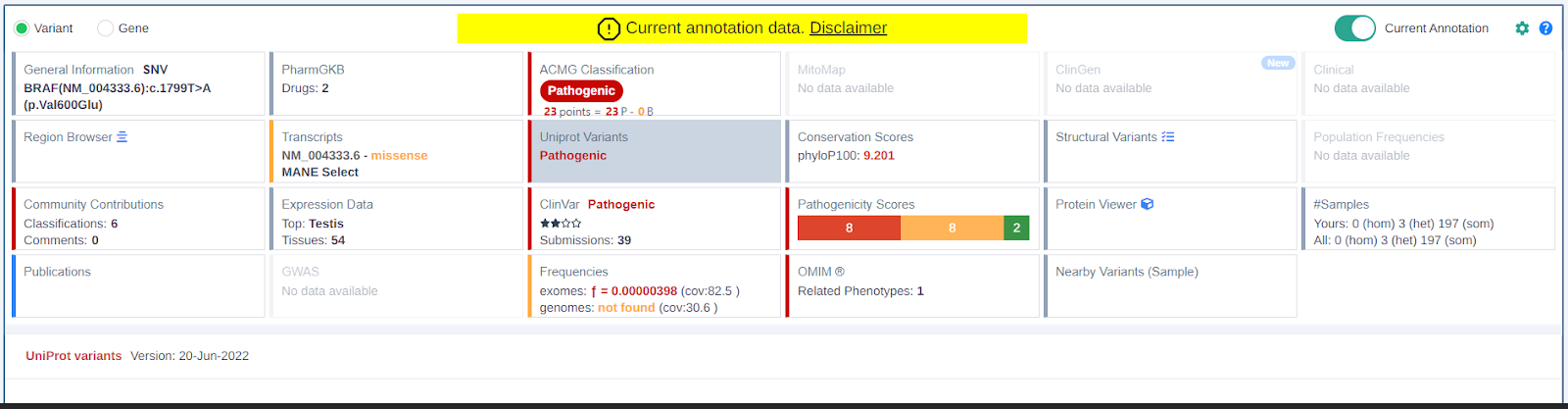
Cards View
Following the introduction of data being displayed in a 'card view' we have received feedback requesting an even more “Compact” version. This can be selected by selecting 'Compact View' in the settings option:
Launch CNV Analysis from VarSome Clinical API
It is now possible to launch a CNV analysis using the VarSome Clinical API. Details on how to use this new feature can be found in the VarSome Clinical API documentation.
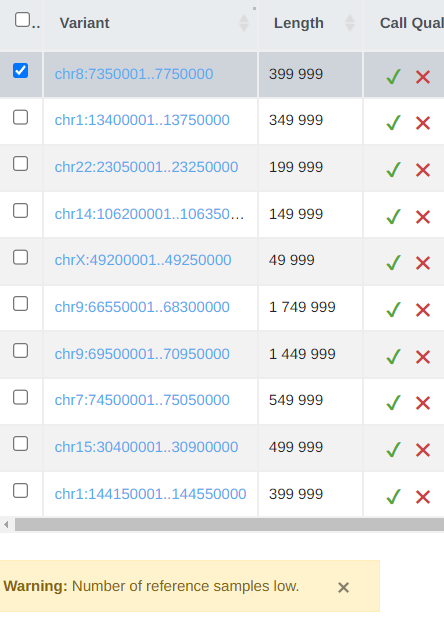
CNV Analysis Call Reliability
Although we highlight the quality score of the CNV call in the QC report we also now display a number of warnings on the variant table.
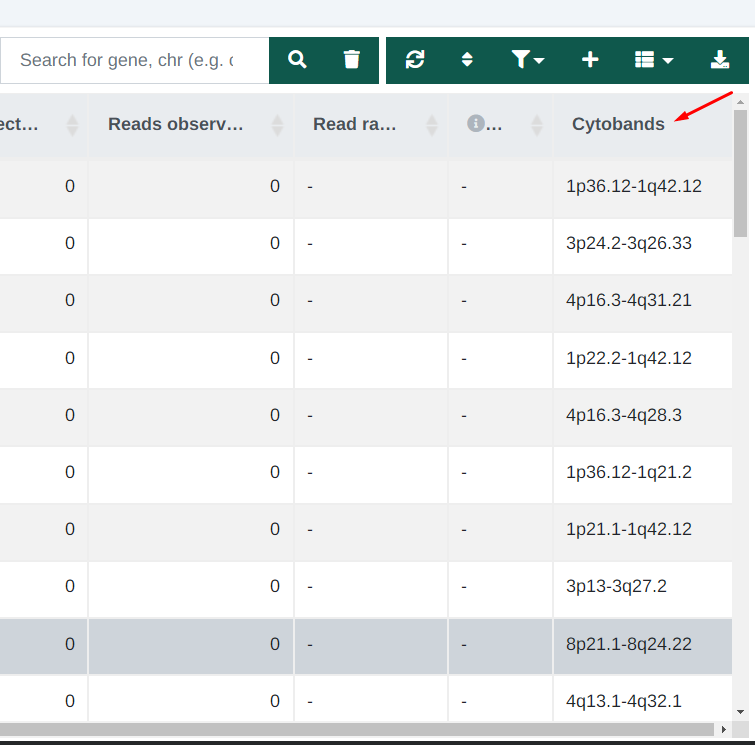
CNV Analysis - Cytoband column
An additional column has been added to the CNV variant table that shows the CNV cytoband information:
CNV Analysis - Combined SNPS and CNVs warning message
We now show a warning message that alerts you to the fact that when you are viewing an SNP there is also an associated CNV or deleted gene. We display 3 new messages:
- No CNV Overlap:
- This variant has an overlap with a CNV:
- An error has occurred and cannot determine if there is a CNV overlap:



Filter - Allele Frequencies
In order to avoid confusion with the naming of the frequency filter we have renamed the “Allele frequency filter” to “Population frequency”

ACMG Classifier
We have implemented most of the ClinGen standards & guidelines for mitochondrial variants:
- Specific frequency thresholds are used for rules BA1, BS1 and PM2
- Rules PM1, PP2 & BP1 are disabled
- Two specific in-silico scores are now used: MitImpact for missense variants, and MitoTIP for non-coding variants
- Rule PVS1 does not attempt to evaluate nonsense mediated decay (NMD), as it is not known to occur for mtDNA
- Rule BP7 now applies to all synonymous mitochondrial variants
- We have improved how MitoMap entries are interpreted as 'clinical evidence'
- “Confirmed” variants are coined as 'Pathogenic'
- “Reported” variants are counted as 'Likely Pathogenic'
- variants with frequency >= BS1 cut-off are classified as 'Likely Benign'
- variants with frequency >= BA1 cut-off are classified as 'Benign'
- All other variants are now counted as VUS
We will continue refining this implementation in our next release.
We also made some minor improvements for missense variants:
- The classifier now uses MetaRNN instead of BayesDel_addAF.
- The threshold for rule BP1 has been recalibrated for improved specificity.
VarSome
Region Browser
We are very proud that this release sees the launch of our new GenomeBrowser.
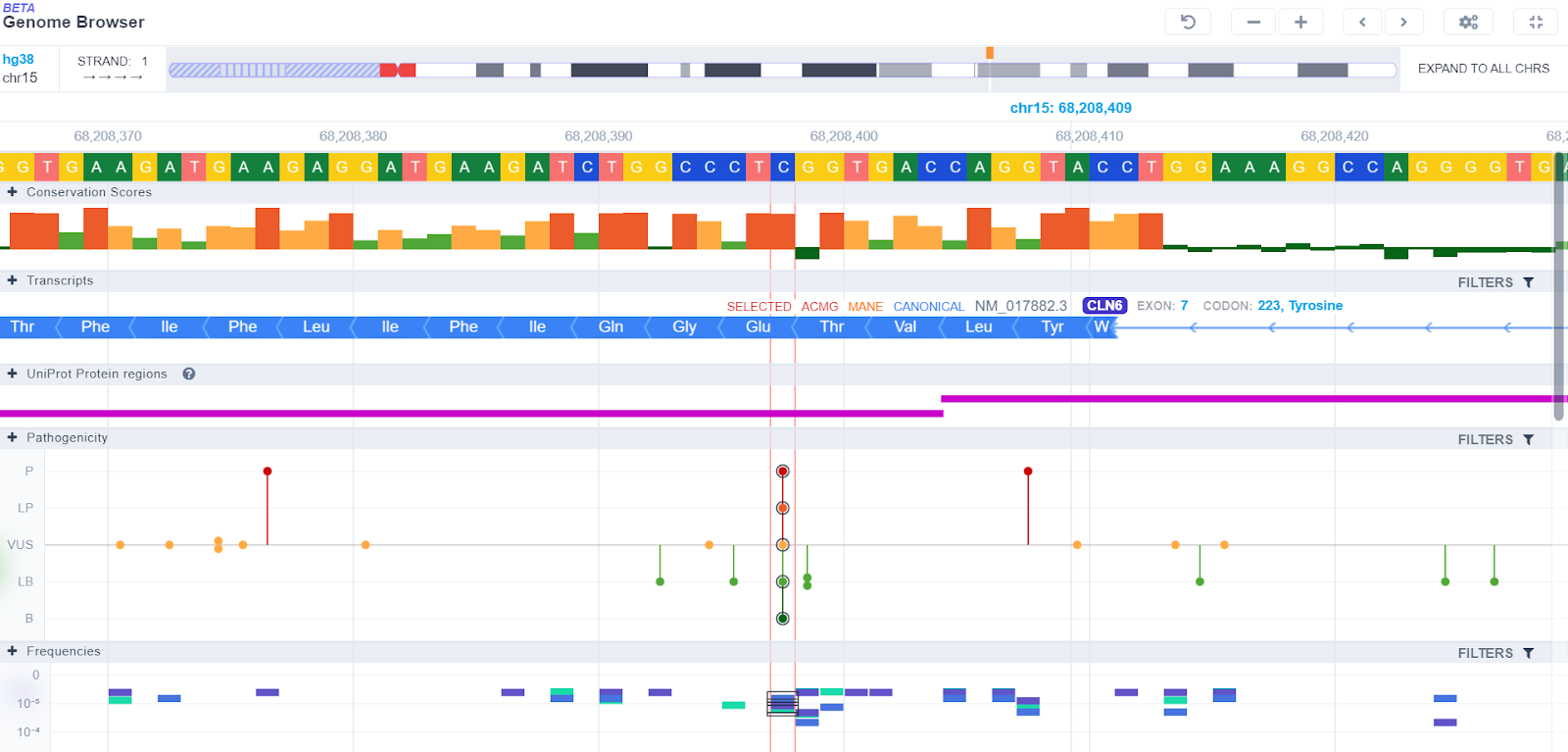
The new browser is quicker to load and easier to navigate. It also introduces key new features:
- New Conservation score track
- Better filtering
- The ability to visualize the whole chromosome
- The ability to reverse the strand
Over the next few months we will be making further improvements.
If you have any suggestions or experience please let us know. You can provide feedback using the option on the top right corner of the browser.
If you would like to go back to the legacy browser you can click the link on the top left hand corner
For further details on the features and best way to use the new browser please refer to the Region Browser User Guide.
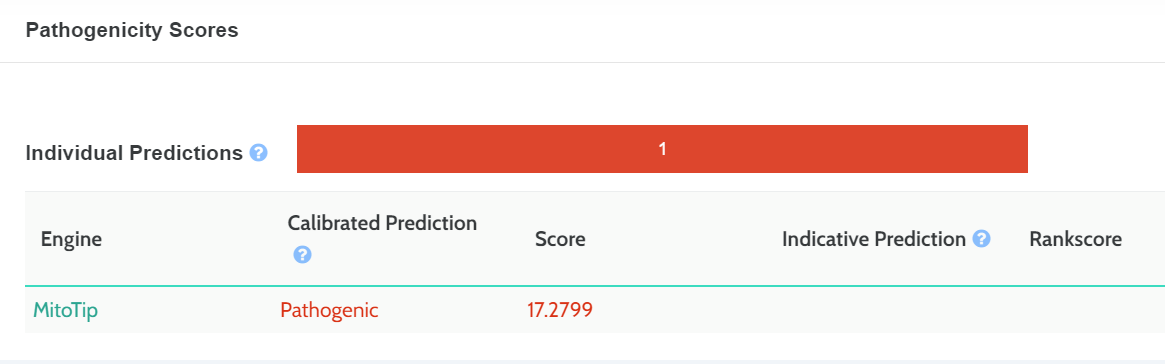
In Silico Predictions
MitoTip and APOGEE pathogenicity scores have been add to the in-silico prediction scores:
Components
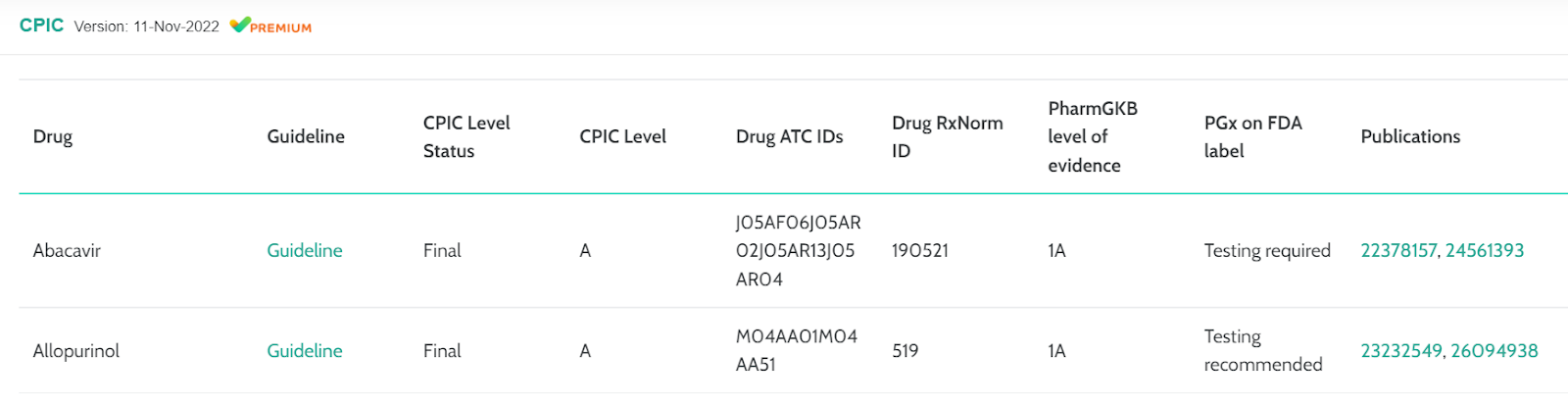
Clinical Pharmacogenetics Implementation Consortium (CPIC)
CPIC is a data source designed to help clinicians better understand how to optimize drug therapy. Recently they have added several new fields which we now display in VarSome Premium:
- CPIC level status
- Drug ATC IDs
- Drug RxNorm ID
Further Information and Support
Not already a VarSome Premium, VarSome API or VarSome Clinical user? Get in touch and ask for a free trial.
As ever we hope you find these changes and improvements helpful, we’d love to hear any suggestions you may have, support is available as usual from support@varsome.com
- The VarSome Team
Submit a Comment