
Aggeliki Karabassi
CTO
Summary of Key New Features for 11.8
Two-Factor Authentication added as an additional security layer across VarSome, VarSome Premium, and VarSome Clinical
VarSome Clinical
- New Sample table format
- Upload BAM files with associated VCF files
- New public gene lists for Secondary findings genes
- Filestore 30-day retention
- VarSome Picks additions
- Improved PGx report
- Support MGI file names
VarSome Classifier Changes
- Curated publications by the VarSome curation team
- ClinGen recommendations for the BA1 rule
VarSome
- LOVD - added genes component (VarSome Premium only)
- DVD component integration
- Frequency component - added gnomAD Hemizygous data
- In-Silico predictions score card renamed In-Silico Engines
- ClinVar comments data now included from contributions
NOTE: For VarSome Premium users, in September as part of the 11.9 release we will be removing the data source PolyPhen.
New Features
Two-Factor Authentication
For an additional layer of security we have provided the option to sign in using 2FA.
Three sign in options are available:
- SMS
- Authenticator app
- Generation of backup codes
Additionally the 'remember this device' option is provided on your browser for 30 days after initial successful login.
For further information please see our related 2FA Help Page
VarSome Clinical
New Sample Table Format
Samples information is now displayed in tabular format with all sample data appearing on one line.
Upload BAM Files with Associated VCF Files
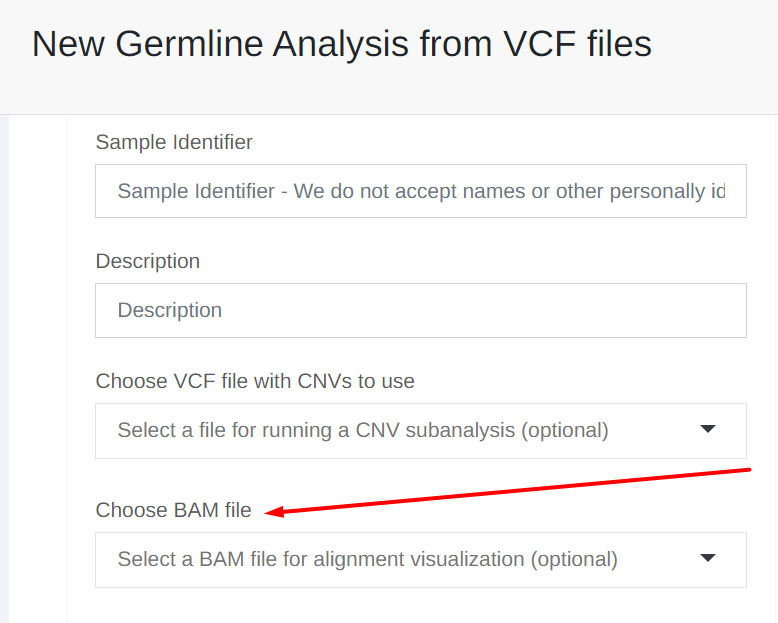
Users starting their analyses from a VCF file can now optionally upload BAM files for alignment visualization. Whenever a user adds a BAM file, we will display links to IGV and JBrowse in the 'Coverage' column of the variant table. These links will allow the users to visualize the alignments in each variant position. (Please note that the alignment visualization is already available to users starting their analyses from FASTQ files).
Once uploaded you can select the BAM file to associate with the VCF file from the 'New analysis from VCF files'.
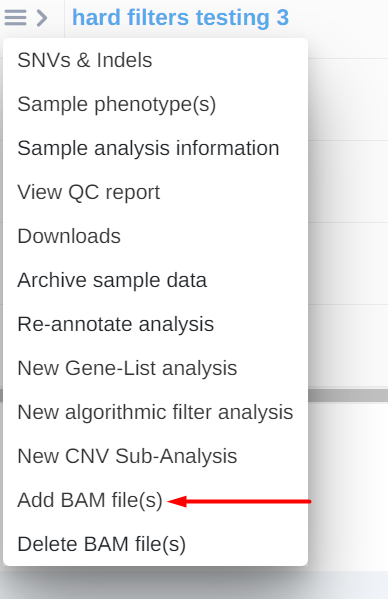
Or
From the analysis actions menu so you can also use the option once the analysis is done.
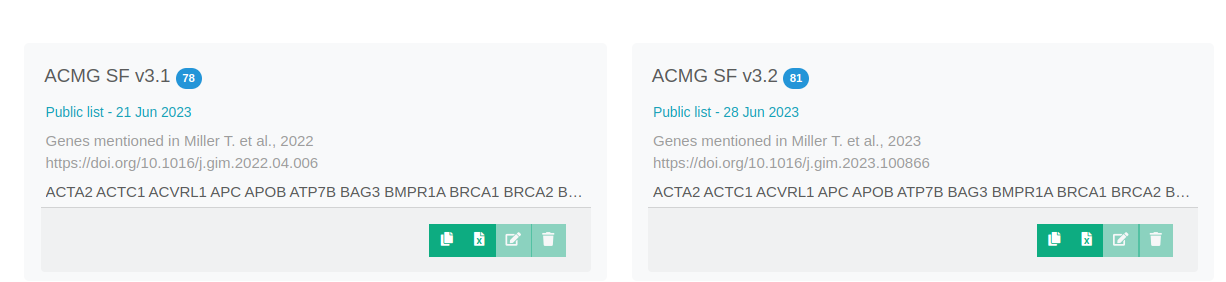
New Pubic Gene Lists For Secondary Findings
We have added two new public gene lists containing the genes mentioned in the two latest versions of the ACMG Secondary Findings Guidelines (versions 3.1 and 3.2).
Filestore 30-Day Retention
All uploaded files that have not been used to launch an analysis within 30 days will be automatically deleted.
VarSome Picks Additions
VarSome Picks can now run for FASTQ files as well as from VCF against hg38 and hg19 reference genomes for the selected assay for germline and or gene list analysis.
The following default options will now be used and run automatically for VarSome Picks:
- The algorithmic filter will run automatically ONLY when phenotypes are provided.
- The algorithmic filter will re-run automatically whenever phenotypes are added (even if not initially provided) or changed from the user in an existing analysis. Every time the filter runs, the previously produced output will be erased and a new one will be produced.
- If the user does not provide phenotypes, the user will still have the option to run VarSome Picks (the algorithmic filter will not be triggered automatically). In this case, a warning will pop up to make sure that the user will be aware of the limitations of the algorithmic filter to produce the expected results.
Warning: This sample has no associated phenotypes. While the VarSome Picks filter can still run, the results will be less informative when the sample’s phenotype(s) are unknown.

PGx Report
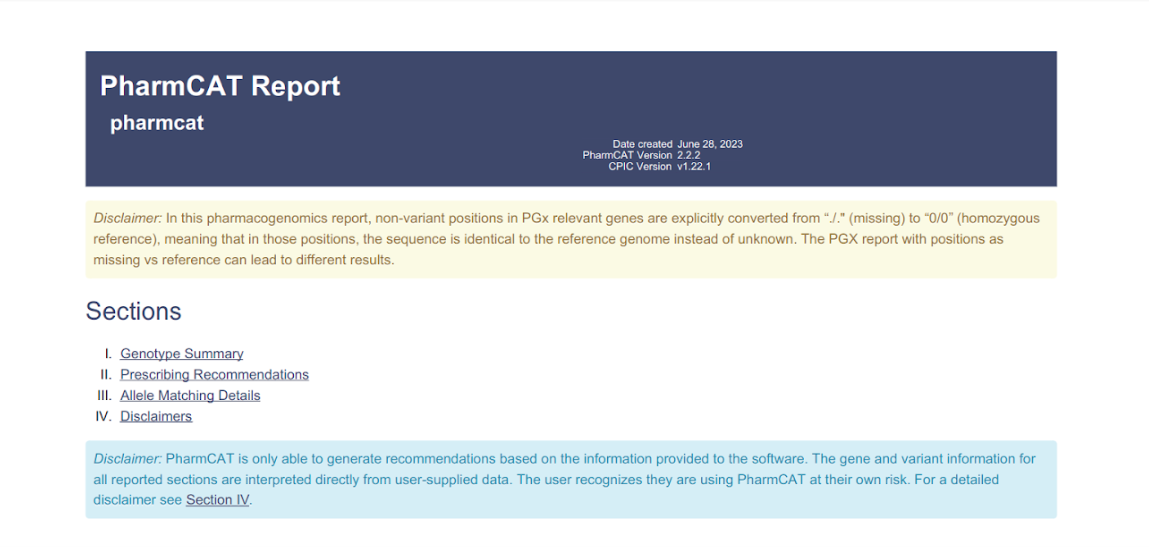
Typically in WGS samples, a large fraction of the PGx relevant positions that are considered by PharmCAT are absent from the VCF input. If those missing positions are assumed to be 'no-call', the resulting PGx report results in the output of multiple possible genotypes, and hence less specific treatment recommendations.
In VarSome Clinical, we assume that all missing PGx relevant positions are homozygous reference. However, this may not reflect reality, because such positions may in fact be unreadable or uncallable. Running PharmCAT with positions as missing vs reference can lead to different results. We have added in the PGx report a disclaimer (in yellow) about the default parameters we use to run PharmCAT Annotation tool, as shown below.
Support MGI File Names
We now support MGI file names with the pattern [flow cell ID]_[lane ID]_[barcode ID]_(optional id)_[read 1/2].fastq.gz and we group them with sample name [flow cell ID]_[barcode ID]:
For further information see: https://en.mgi-tech.com/
Help and Support
We have renamed the Support menu to 'Help'. Here the users can download the VarSome Clinical user manual in PDF version, they can access the VarSome Help Center to find answers to specific questions, and they can also request a new assay to be added.
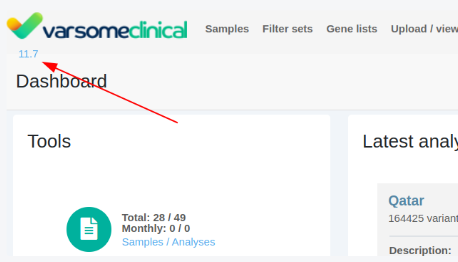
Version Number - Link to Release Note
The current software version number is displayed. By clicking on this version number you are directed to the related release note version.
VarSome Classifier changes
Curated Publications by the VarSome Curation Team
Our experienced curation team work at linking publications and classifying variants that are currently showing as variants of uncertain significance.
Curated entries are available only for our paying customers. Any publication linked by our Saphetor curation team is now highlighted in the VarSome publications component.
Additionally, we have increased the importance of the VarSome curated publications in the germline classifier so that VarSome curated entries have more strength as they may trigger the PP5/BP4 and/or PS3/BS3 ACMG rules.
ClinGen Recommendations for the BA1 Rule
Following the ClinGen SVI expert panel recommendations for the Benign Stand Alone ACMG/AMP Criterion, our classifier is now excluding the frequencies from the Ashkenazi Jewish and Finnish populations in the BA1 rule.
VarSome
LOVD Gene Card (VarSome Premium Only)
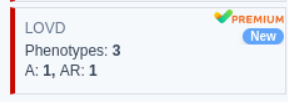
We have added a new gene card containing a summary of phenotype and inheritance information per gene from the LOVD database. This card is available only to VarSome Premium and VarSome Clinical users.
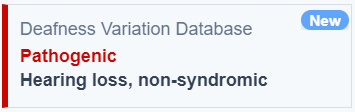
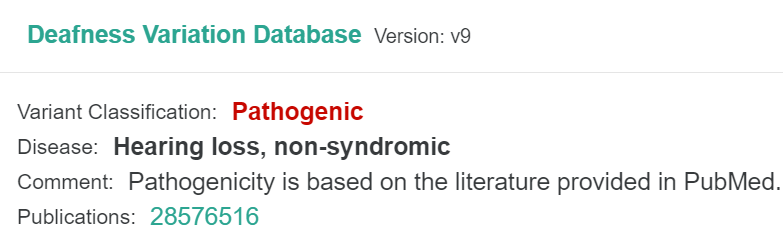
Deafness Variation Database
The Deafness Variation Database (DVD) has been integrated into all VarSome products. This data source collects data from public resources. It provides annotation and classification for genetic variants related to syndromic and non-syndromic hearing loss. The data is curated by experts on hereditary hearing loss.
Frequency Data
Hemizygous Frequency data from gnomAD is now displayed in the frequency table and as a graph if the data is available to display.
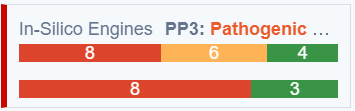
In-Silico Score Card
The 'In-silico Predictions' card has been renamed to 'In-silico Engines'.
In order to more clearly present a pathogenicity summary we have also added to the card the Meta Scores bar.
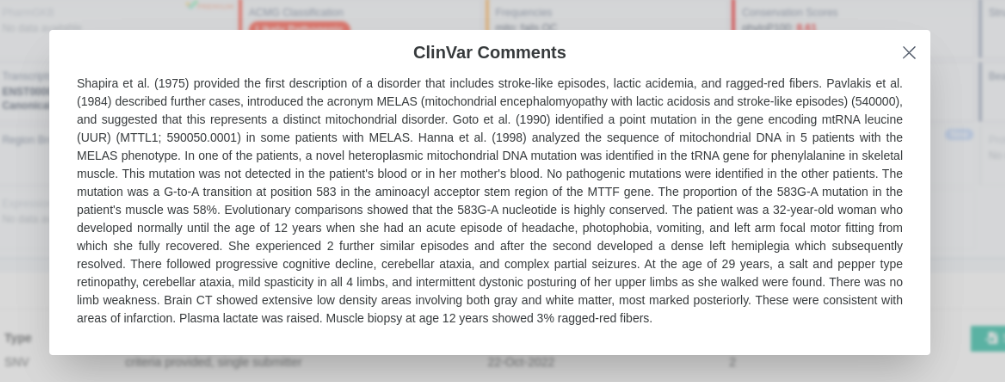
ClinVar
'Comments on submission' data has been included in the ClinVar data displayed.
Support
As ever we hope you find these changes & improvements helpful, we’d love to hear any suggestions you may have, support is available as usual from support@varsome.com.
The VarSome Team.
Submit a Comment