
Richard Meyer
CTO
Our June release introduces a wide variety of enhancements and new features to our platforms.
Highlights
VarSome Clinical Key Updates
- New variant caller “VarDict” for Amplicon samples
- Parameterisable algorithmic filters.
- Additional columns in our exported Excel file.
- New Consanguinity Metrics on Main analysis.
VarSome Key New Features
- New Database of Curated Mutations (DoCM)
- 3 x New gene databases
- EVE pathogenicity score for missense variants
Continual Improvements
ACMG classification:- Amended PVS1 rule to include NMD prediction for missense and frameshift variants
- BS2: reduced threshold for homozygous to 1 (was previously 2).
- PP3: will no longer be applied if rule PM4 was triggered.
- BA1: will now always trigger a benign verdict even if there is strong conflicting pathogenic evidence.
- PP5: will now trigger if ClinVar reports “Uncertain Significance” but there are pathogenic submissions.
- PP1 and BS4 will now trigger if the “Family Segregation” field is provided in the “Optional Sample Information” in VarSome Clinical. PP1 will trigger if it is set to “Yes”, while BS4 will trigger if it is set to “No”.
- The Database of Curated Mutations (DoCM) has now been integrated into our AMP classifier in the curated evidence rule.
- ACMG rule PVS1 is disabled for Oncogenes.
- The inheritance rule will adapt accordingly if the “Family Segregation” field is provided in the “Optional Sample Information” in VarSome Clinical.
- The accuracy of the CNV classifier has increased, as a result of improvements in the overlap rule.
- Faster CNV annotation times
- Added cytobands for CNVs
New Features
VarSome Clinical
Variant Calling
As previously mentioned in the 11.3 Pre Release note, this release affects variant calling for samples using Amplicon.
Specifically, we are moving to a new variant caller, VarDict, for Amplicon samples only. Our testing has shown that this caller performs better on sequencing data produced by Amplicon-based assays. This new version of VarSome Clinical will be using VarDict for all Amplicon samples.
No change will be made to capture-based samples or any other aspect of the variant calling process.
Parameterizable algorithmic filters
A key new feature of this release is the introduction of parameterizable algorithmic filters.
For several of the existing filters we now provide the option to change specific parameters that previously were fixed. In this way you can customize these filters according to your needs.
For some of the filters, in order to add the parameterization functionality we have integrated similarly functioning filters into one. For example, you can now find, under the filter “Segregating Variants”, the filters previously known as:
- Segregating Variants (dominant, all VUS),
- Segregating Variants (dominant, strong VUS)
- Segregating Variants (recessive, all VUS)
- Segregating Variants (recessive, strong VUS)
- Compound Heterozygous Segregating Variants (all VUS)
- Compound Heterozygous Segregating Variants (strong VUS).
To find them please go to Algorithmic filter analysis. You will see next to certain filters a light blue “Options” button:

After you choose from the drop-down the analysis to which you wish to apply one or more filters, you then need to select the filters by clicking the relevant checkboxes, which will activate the options box.
This box displays the default parameters that we use for this particular dynamic filter.

For further details on the algorithmic filter and the provided parameters please click on the info icon next to the “Options” button.
Once the analysis has run, you can view the filter options selected for the algorithmic filter analysis both in the sub-analysis name

and in the “Sample/Analysis Information” option of the “Analysis Actions” menu:
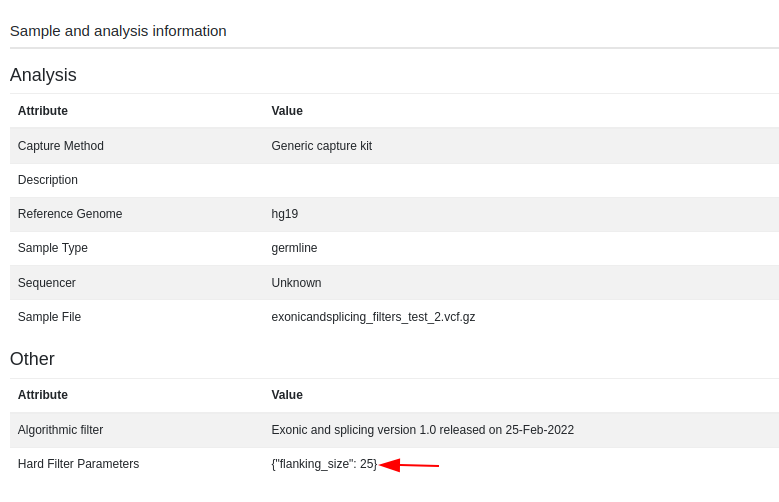
Consanguinity Metric
We now report the ratio of homozygous to heterozygous variants in the sample information screen. This can be used as a measure of consanguinity.

Excel Download
We have added four new columns to the Clinical downloadable Excel spreadsheet:
- Variant functions per transcript used for ACMG
- Alternate allele coverage (allelic balance x coverage)
- Total number of phenotypes that match (Currently we only show the name of matching phenotypes)
- Filters
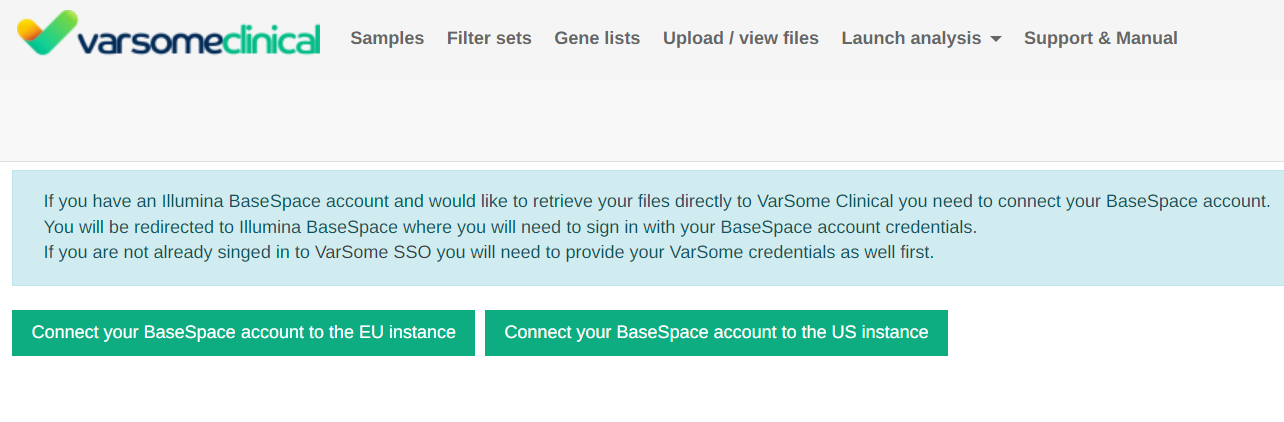
BaseSpace integration
Two options are now available to connect your Illumina BaseSpace account: EU or US. You will need to select the instance where your data is stored.
VarSome Clinical API
You can access the API documentation at https://ch.clinical.varsome.com/api/v1/docs/
VarSome
VarSome Premium - DoCM Card http://www.docm.info/
The Database of Curated Mutations (DoCM) is a centralized highly curated database of curated pathogenic mutations. We now display both the diseases and the relevant drugs from this database if they are available. We also use this data in our VarSome Premium AMP Classification to improve the accuracy of the Curated Variants Rule



Additional Data cards for Genes
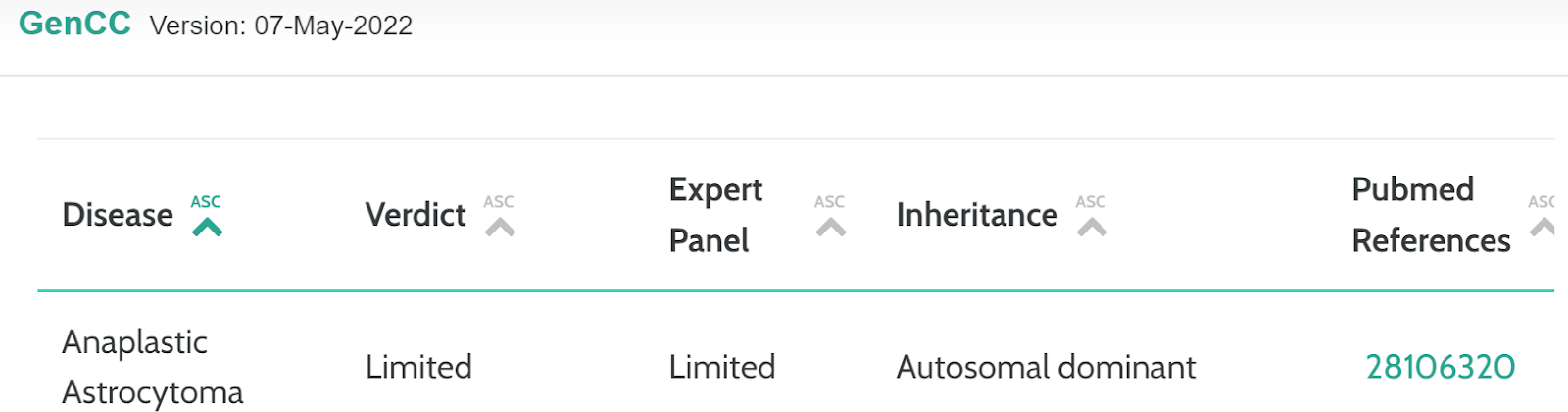
GenCC

Gencc (Gene Curation Coalition) is a database that provides information pertaining to the validity of gene-disease relationships with a current focus on Mendelian, germline disease.
We have incorporated a new card that presents the gene related data and verdict.
NHI ClinGen Gene-Disease Validity

NHI ClinGen Gene-Disease Clinical Validity curation involves evaluating the strength of evidence supporting or refuting a claim that a variant in a particular gene causes a specific disease. We have incorporated a new card on the Gene page that displays verdict of the associated disease

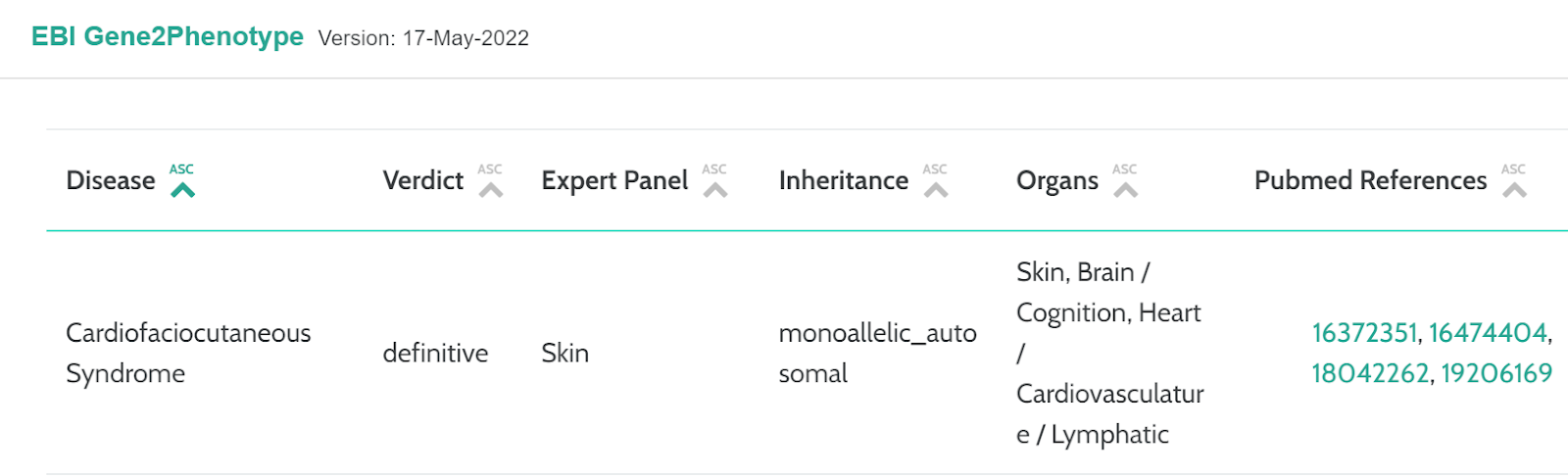
EBI Gene2Phenotype - https://www.ebi.ac.uk/gene2phenotype/

G2P is a publicly-accessible online system designed to facilitate the development, validation, curation and distribution of large-scale, evidence-based datasets for use in diagnostic variant filtering. Each G2P entry associates an allelic requirement and a mutational consequence at a defined locus with a disease entity. A confidence level and evidence link are assigned to each entry. We have incorporated a new card on the Gene page that displays this verdict of the associated disease
dbNSFP - New Mouse Genome Informatics added to this component

Publication Tagging
To avoid confusion we have now merged the tags for diseases and phenotypes and also organs and tissues.

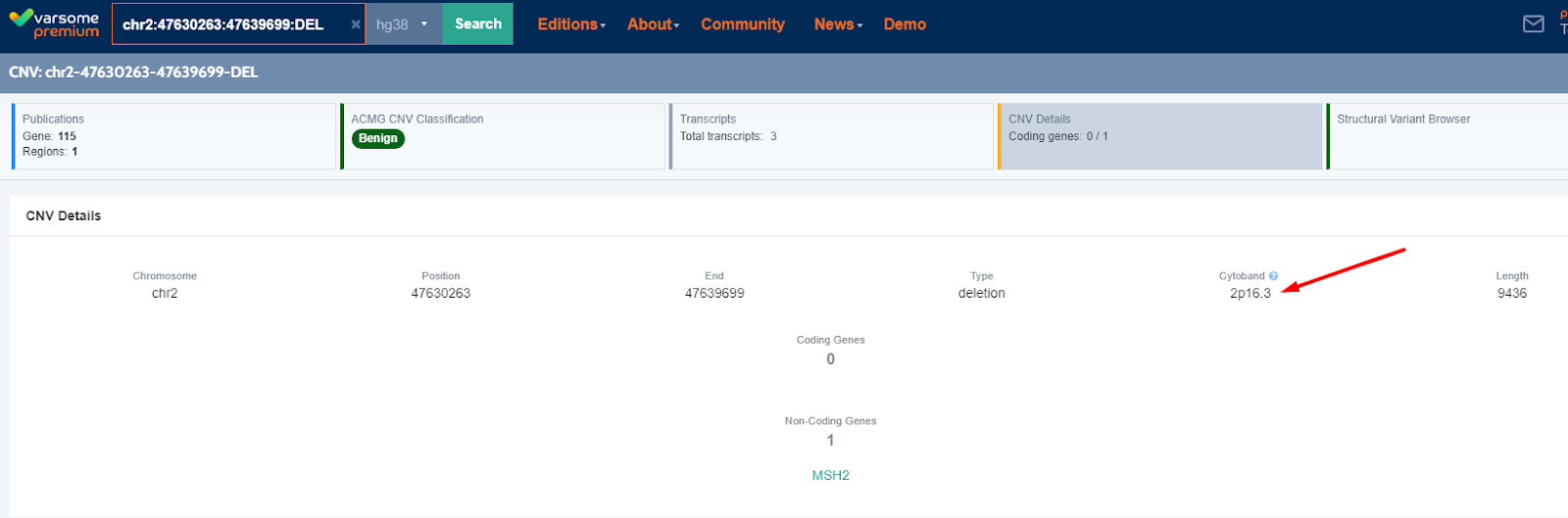
CNVs
In the same way that we show the cytoband (chromosome position) for Single Nucleotide variants we now also show the cytoband associated with the CNV.
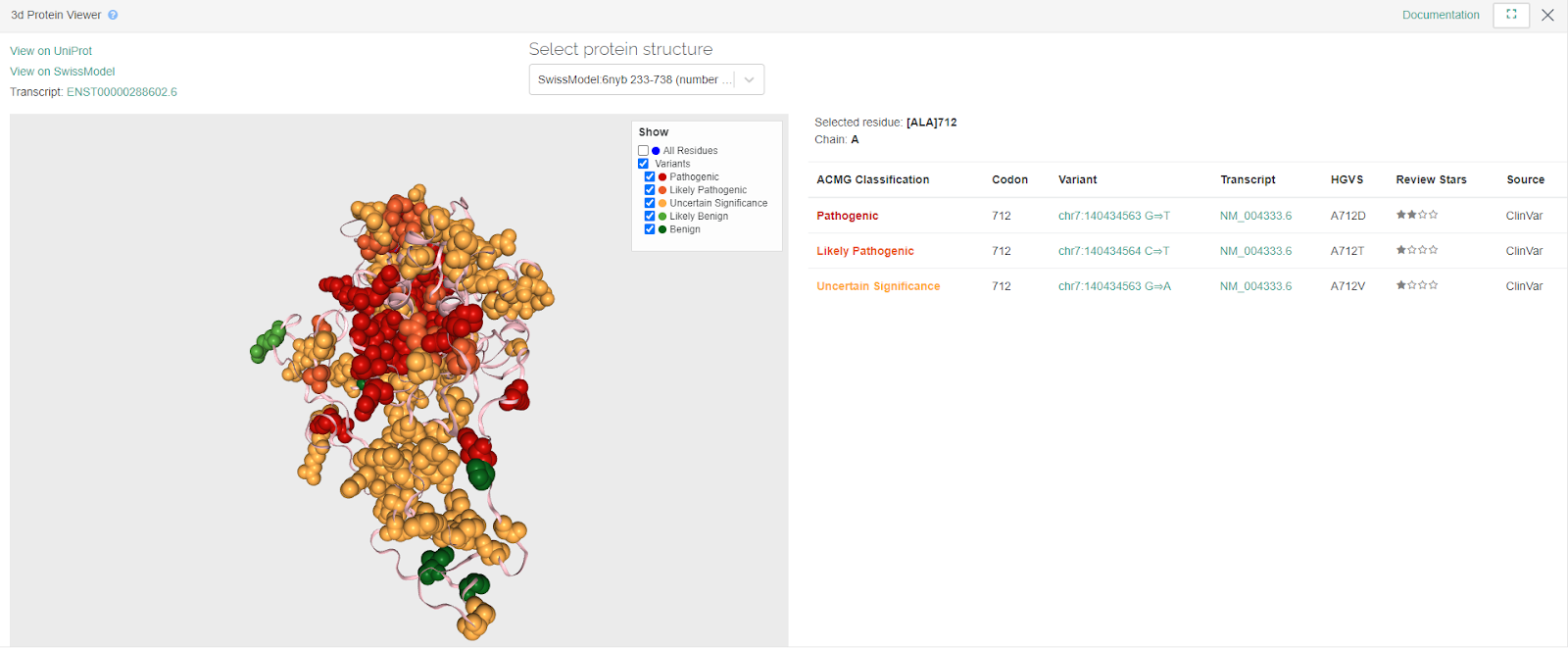
Protein Viewer
The 3D Protein Viewer has been improved so that the related variants are displayed on the right hand side in full screen mode:
Pathogenicity scores
EVE (evolutionary model of variant effect) is a model for the prediction of clinical significance of human variants, based on sequences of diverse organisms across evolution. It uses fully unsupervised deep learning trained on amino acid sequences of over 140K species. Based on this it makes a prediction of pathogenicity for all single amino acid variants of disease related genes. This is represented in the form of a score that ranges from 1, most pathogenic, to 0, most benign.
We now present this score and incorporate it into the pathogenicity score.
For further detail and a more detailed description of the score please see https://evemodel.org/


On linking to VarSome from another application such as ClinVar it is now mandatory to select a reference Genome in order to annotate your variant. We encourage you to use hg38 whenever possible.

Classifiers
As with every release we continue to build on the strength of our automated classifiers following the ACMG & AMP guidelines. As a result our automated classifier is both more accurate and more compliant with the ACMG guidelines and subsequent revisions.
ACMG
We continue to improve our automated ACMG classification, based on user feedback and ClinGen recommendations:
- Amended PVS1 rule to include NMD prediction for frameshift and missense variants. This is done by implementing the following ClinGen recommendations, and specifically this diagram. Specifically, for frameshift and missense variants that cause NMD we take into consideration the reported pathogenic LOF variants in the truncated region and the functional domains that are reported by UniProt in the exon and the truncated region. For frameshift and missense variants that don’t cause NMD, we additionally use the percentage of the protein that the variant removes. In future we also aim to role this out for other variants.
- BS2: reduced threshold for homozygous to 1 (was 2 previously).
- PP3: will no longer be applied if rule PM4 was triggered.
- BA1: will now always trigger a benign verdict even if there is strong conflicting pathogenic evidence.
- PP5: will now trigger if Clinvar reports “Uncertain Significance” but there are pathogenic submissions.
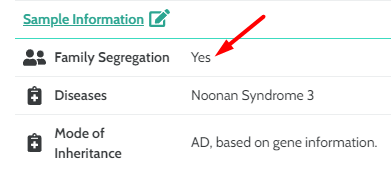
- PP1 and BS4 will now trigger if “Family Segregation” field is entered in “Optional Sample Information” . NOTE: This is also incorporated in the CNV classifier.

If “Family Segregation” is set to “yes” and the entered disease is linked to the variant’s gene then PP1 will trigger.
If “Family Segregation” is set to “no” and the entered disease is linked to the variant’s gene then BS4 will trigger.

BS3 - Up until now when a user classifies a variant it would only be classified on the reference genome browser they were using. So if you looked at the variant in the other reference genome it would not be considered in our classifiers. We have now lifted over any user supplied classifications form either hg19 to hg38 or vice versa depending on which was linked so that we can use it in the classifier, regardless of which genome was used.
We highlight when a classification has been lifted over.
CNV
Increased accuracy by improving the Overlap rule, which now takes into account more conditions, such as the number of pathogenic LOF variants contained in the region and the breakpoints of the provided CNV.
Added “Family Segregation” in the “Optional Sample Information” which will impact the inheritance rule in case the sample’s phenotypes or diseases are matched to the affected regions’ ones.
Improved annotation speed, it could perform up to 10 times faster for CNVs more than 10MBases long.
Further Information and Support
Not already a VarSome Premium, VarSome API or VarSome Clinical user? Get in touch and ask for a free trial.
As ever we hope you find these changes and improvements helpful, we’d love to hear any suggestions you may have, support is available as usual from support@varsome.com
- The VarSome Team
Submit a Comment