
Richard Meyer
CTO
Our February release introduces a wide variety of enhancements and new features to our platforms.
Highlights
Key New Features
- 3D Protein Viewer:
- Single Reads visualization
Continual Improvements
- CNVs: improved use of phenotypes, enhanced display, ability to upload a single VCF file containing both CNVs and smaller variants.
- ACMG classification: improved accuracy & closer adherence to standards.
- AMP Drug rule: now includes approved drugs from EMA in addition to FDA.
Clinical Updates
- Ability to run FASTQ analysis in targeted/non-targeted mode.
- CNV analysis containing SNPs will now generate two separate files.
For additional information on the Clinical platform updates please refer to the 11.2 version User Manual.
3D Protein Viewer - New Feature
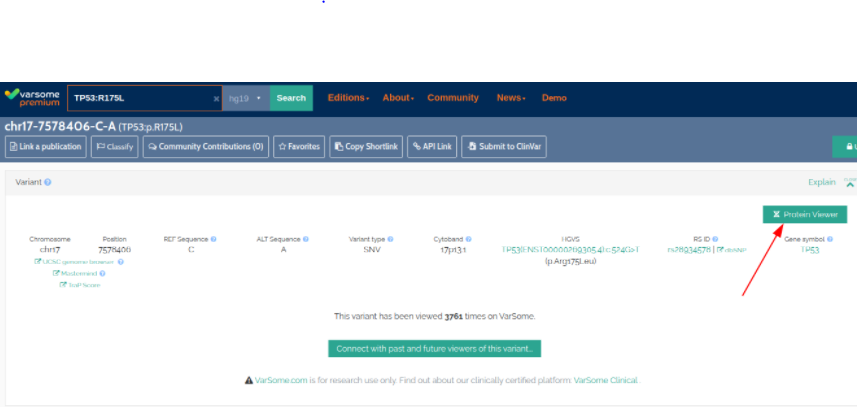
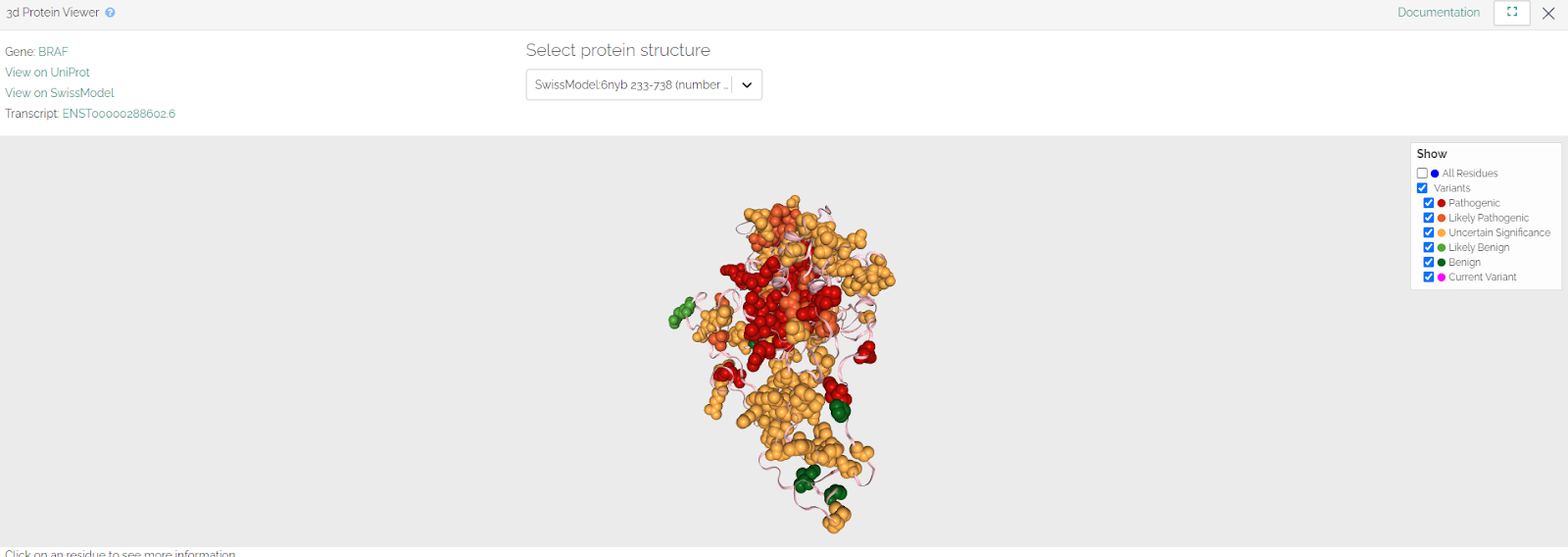
We have introduced a new 3D Protein Viewer. This tool enables you to map variants onto a protein structure.
Details on how to use this new feature can be found here: https://docs.varsome.com/en/protein-viewer
To access the 3D Protein Viewer click on the “Protein Viewer” button on the top right of the basic variant information page, or top right of the gene page.
Single NGS or Sanger Read Visualization - New Feature
You can now search on single NGS or Sanger reads in VarSome. The application will scan the entire human genome to find matches for the sequence entered and list any variants it may contain and their HGVS notation if available.
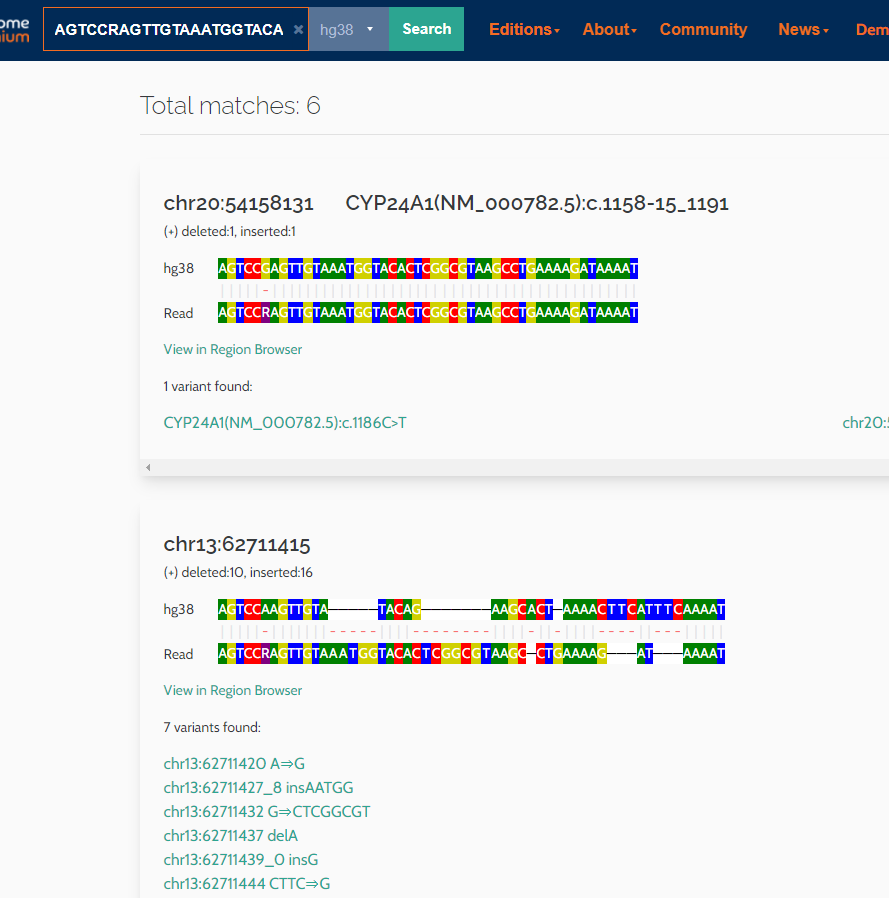
Examples have been added to the help text to show how the single read query should be formatted:
Publications
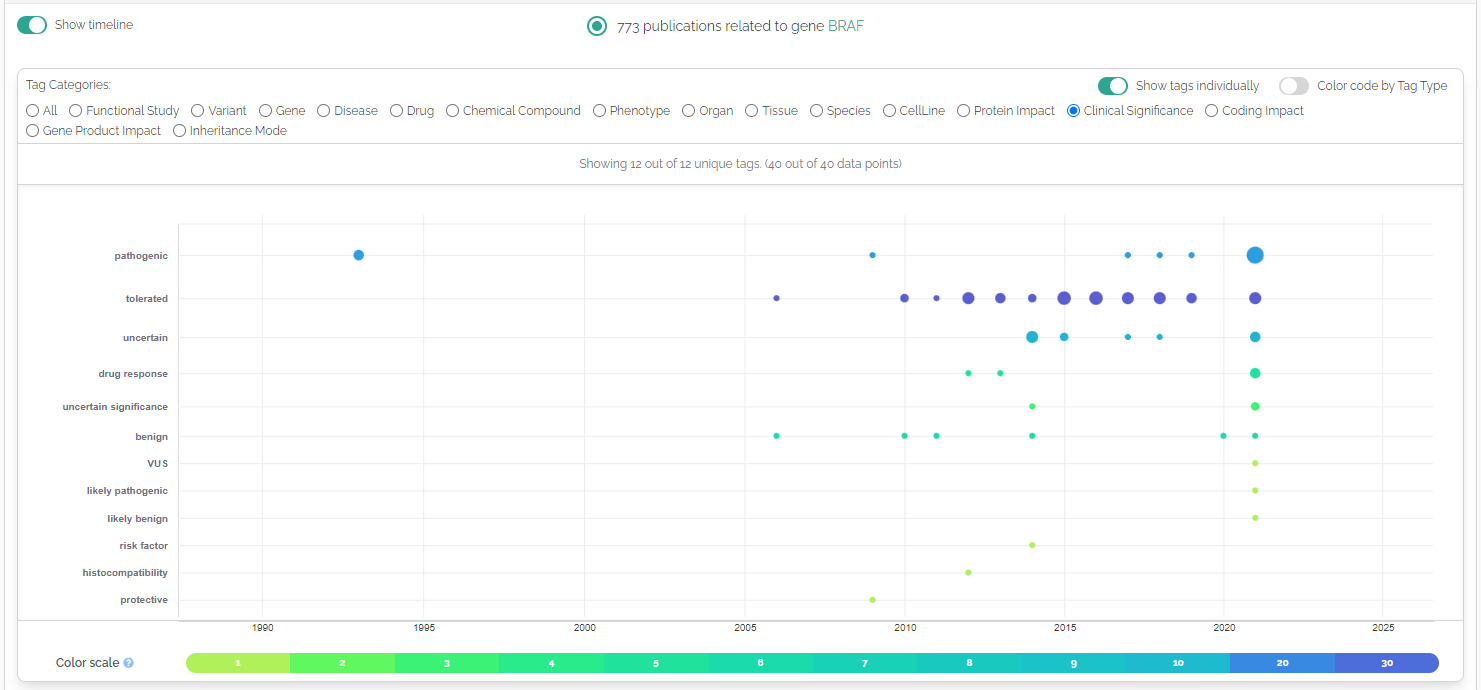
Additional tagging categories have been added to the publications component:
- Inheritance mode: identifying terms such as dominant, recessive, X-Linked etc.
- Clinical significance: matches ACMG classifications or other indications of pathogenicity.
- Coding Impact: terms such as missense, nonsense or cryptic splice-site
- Protein Impact: misfolding, cellular stress etc. affecting the protein or peptide.

VarSome Improvements
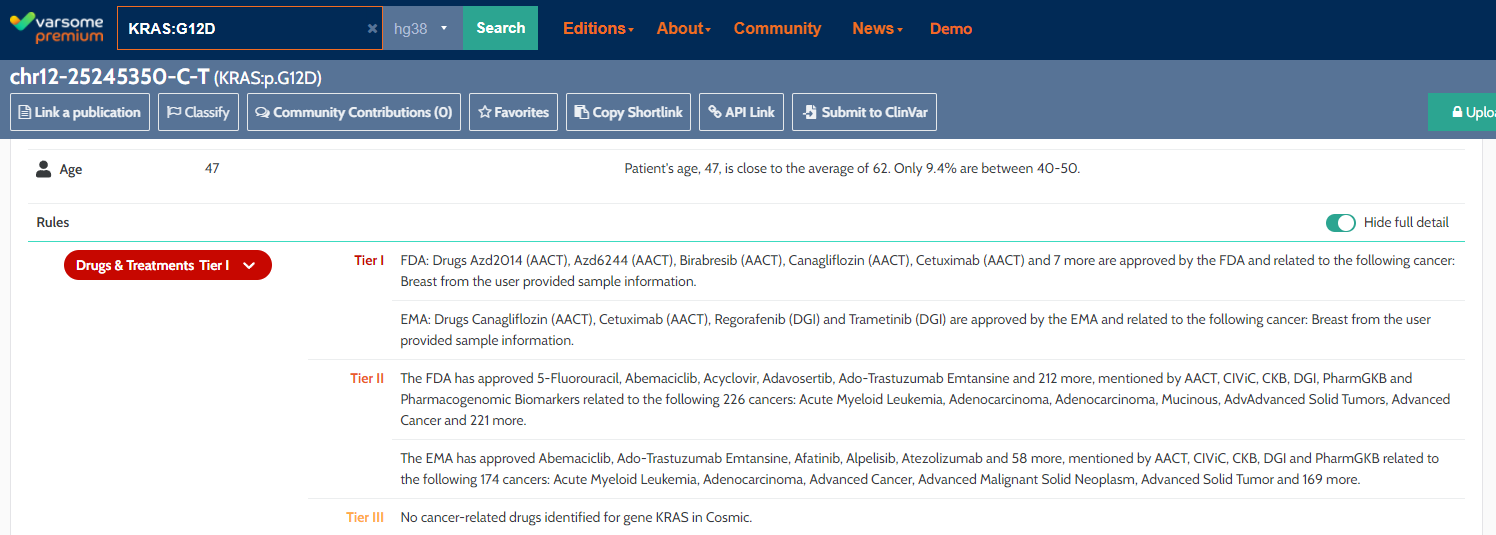
AMP Enhancements:
We have expanded and improved our AMP Drug rule to include approved drugs from EMA in addition to FDA. They are reported separately in the explanation of the Drug rule.
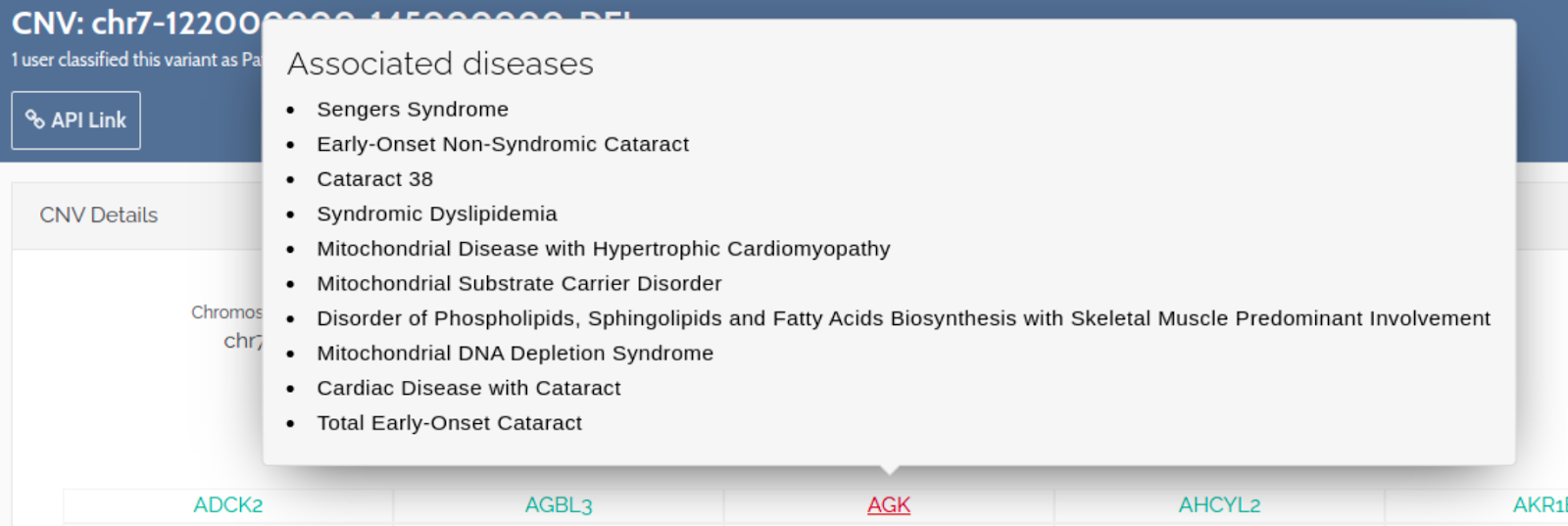
CNV Enhancements:
On the CNV gene table, we now display pathogenic genes as red and display a list of the gene associated diseases.
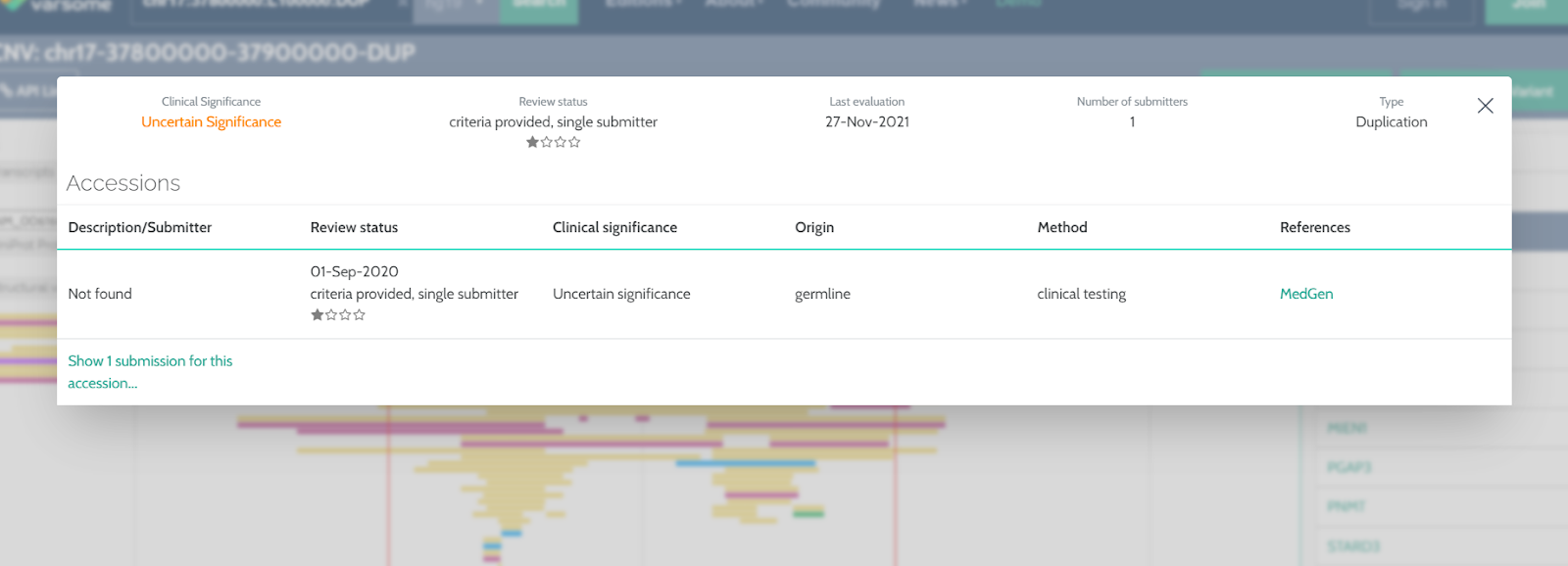
ClinVar CNV regions
We now provide a link to “ClinVar data” on the CNV region browser which allows you to visualize the ClinVar component with information about that particular region

CNV Phenotypes
As with SNPs, we now request input of phenotype/disease information for CNVs. This information is optional. However, adding patient inheritance information in the “Optional patient information” box will improve the quality of your ACMG classification.
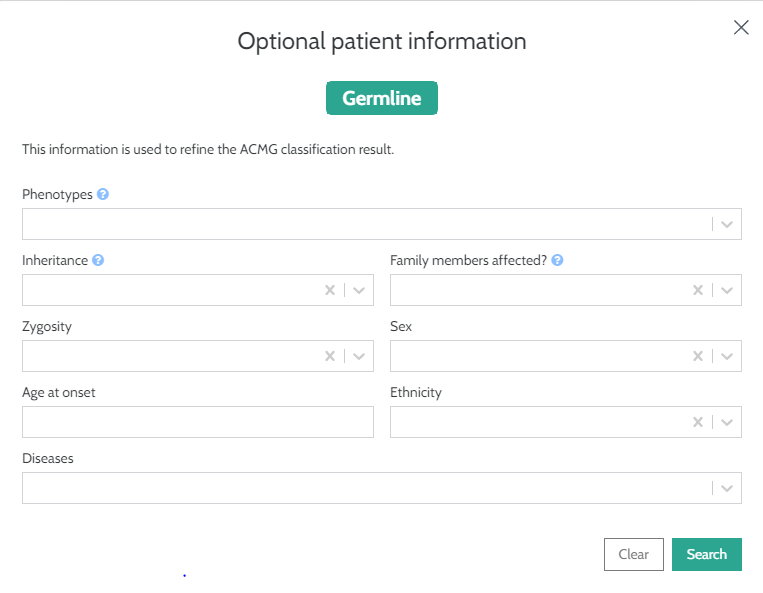
Now instead of seeing the inheritance rule like this:
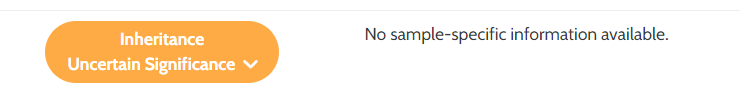
It will be activated:
Classifiers
As with every release, we continue to build on the strength of our automated classifiers following the ACMG & AMP guidelines. As a result our automated classifier is both more accurate and more compliant with the ACMG guidelines and subsequent revisions.
ACMG
- Rules PM2 & BS2, PP2, BP1: the mode of inheritance for a given gene now leverages multiple databases: GHR, Mondo, and ClinGen, furthermore it can use the DOMINO prediction if there is insufficient data from these sources.
- Rule PP3 is no longer triggered if rule PVS1 was triggered, in line with the ACMG guidelines. As a result many variants which were previously annotated as Pathogenic may be downgraded to Likely Pathogenic.
- Rule BP3 will now trigger for missense variants located in a poorly conserved region with no known pathogenic variants.
- Rule PM4 triggers with strength Strong for stop-loss variants that are highly conserved (this is inline with UK guidelines which recommend triggering PVS1).
- Rule BP7 will trigger for non-intronic, non-coding variants that are not conserved and not predicted splicing (this was previously implemented in rule BS1).
- Rule BS1 is a little more conservative: it uses higher thresholds in genes which contain high-frequency pathogenic variants.
- Rule PM1: we have made minor adjustments to the internal thresholds used for hot-spots and domains.
- Rule PM4 now checks that LOF causes disease, similarly to rule PVS1.
- A “Likely Pathogenic” verdict now requires one moderate + four supporting rules, vs three previously (as some rules could not be implemented), in line with the ACMG guidelines.
The sample’s disease information that you enter is now taken into account supplementing the already existing phenotypes matching, triggering the inheritance rules whenever that’s applicable.
AMP
We have integrated the EMA drug approvals into the AMP Drug Rule. You will now be able to see a separate explanation regarding the EMA approved drugs, the same way as the FDA approved drugs.
VarSome Clinical
We have provided the ability to use the “filtered variants” for CNV analyses. It is possible to export all variants on the page to an excel file in the same way as you currently can for SNPs and small INDELs
Filtering
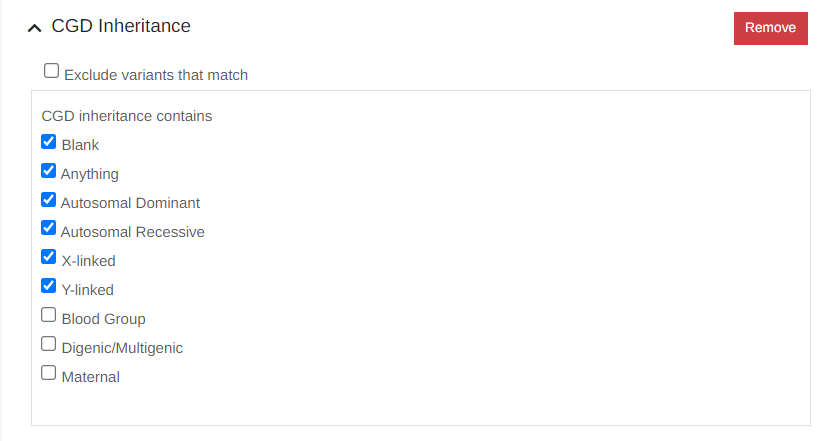
It is now possible to select multiple inheritance modes from a pick-list when filtering:
Dynamic Filters
We have removed the “trash bin” icon next to the “edit” button on the component filter. Now you can no longer accidentally delete an individual component filter when attempting to edit it. We have left the option to delete the main filter set.
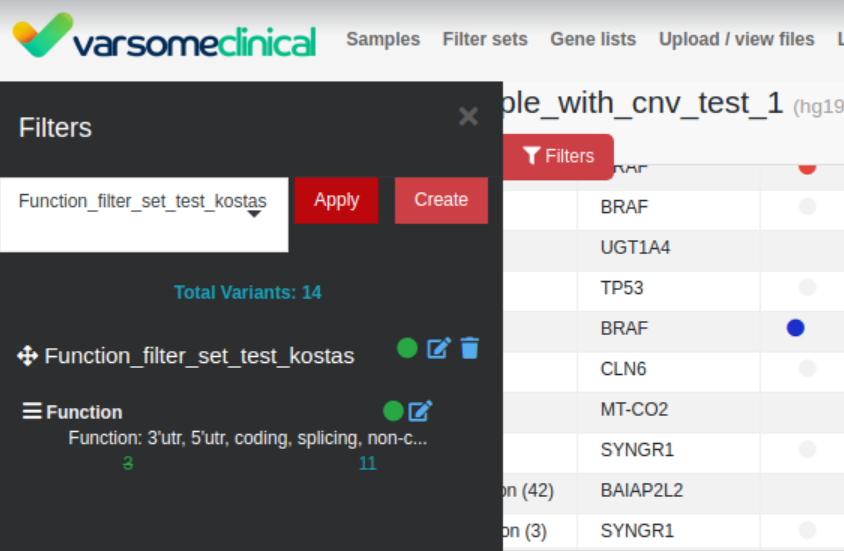
Export to Excel
In addition to data columns for “all samples”, we have now added three new columns for “your samples” in the Excel export:
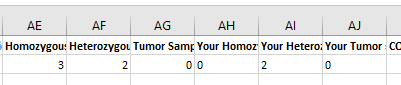
Improve Filtering on Gene List
You can now type a letter to search the gene list dropdown, reducing the need to scroll:

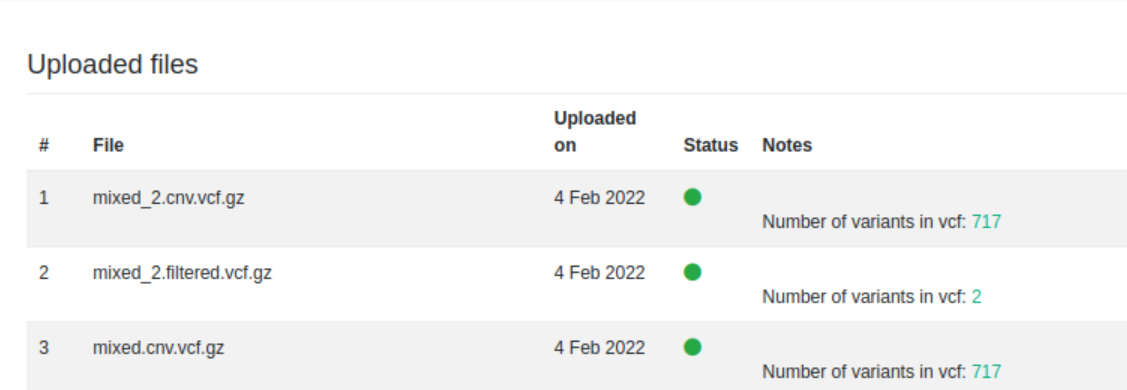
Uploading CNVs and SNPs/indels
We now accept VCF files that contain both copy-number-variants (CNVs) and SNPs/indels in the same file.
As you upload a mixed VCF file, it will now split into 2 files (i.e .filtered and .cnv). For further information about how this works please refer to the user manual.
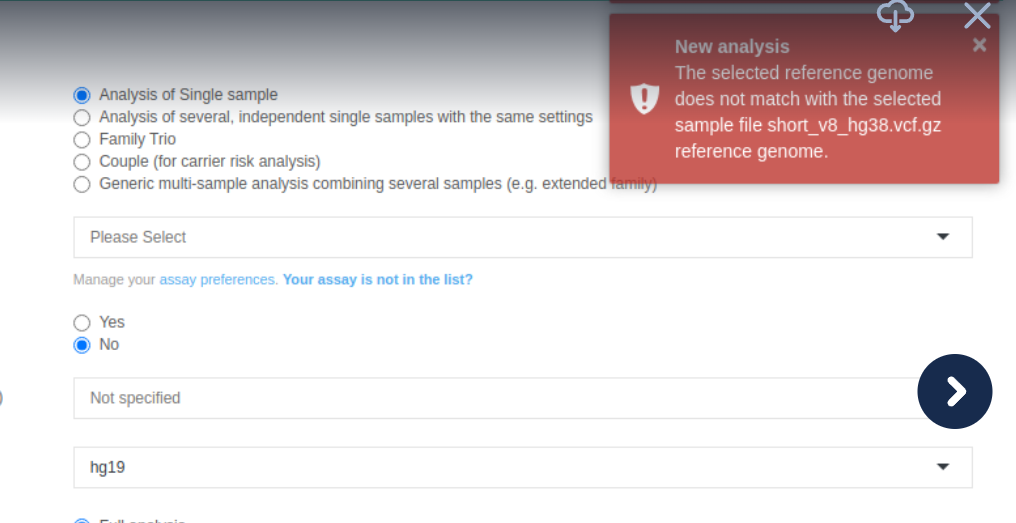
Launch Analysis - Reference Genome
When launching an analysis on a VCF file, we will read the file and attempt to establish the reference genome used in the VCF. If the selected reference genome does not match that in the VCF, we will display a warning message.
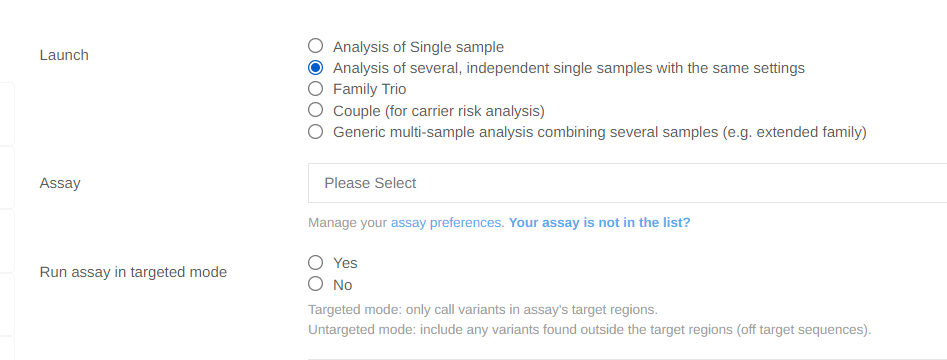
New Targeted/Non-targeted mode for Germline Analysis from FASTQ files:
We have added an option to launch analyses in “targeted” or non-targeted” mode.
Targeted mode will only call variants in the assay’s target regions.
Non-targeted mode will include variants found outside of the target regions if present.
The mode selected will be shown at the bottom of the Sample and Analysis information page
Associate CNV VCF files with component samples of multi-sample analyses
You can now associate a VCF file with CNVs to each component sample of a multi-sample analysis and the CNVs in the file will be annotated when you launch the multi-sample analysis.
VarSome Clinical API
The VarSome Clinical API has been extended to include support for launching germline analyses from FASTQ files (single and multi sample).
Notable changes for analyses from VCF and FASTQ files:
- The sequencer field accepts only one of two values: "Illumina" or "MGI".
- varsome_gene_list_id and panelapp_gene_list_id properties have been renamed to varsome_gene_list_ids and panelapp_gene_list_ids and now expect a list (array) of gene list identifiers. If more than one identifiers are provided the gene list analysis produced will include the combined genes from all gene lists provided
Launching analyses from FASTQ files
The set of parameters that the relevant API endpoint expects when launching an analysis from FASTQ files are similar to the ones provided for VCF files with some differences:
- capture_kit_id parameter is required
- sequencer parameter is required
- capture_kit_targeted_mode parameter is required
You can access the API documentation at https://ch.clinical.varsome.com/api/v1/docs/
Further Information and Support
Not already a VarSome Premium, VarSome API or VarSome Clinical user? Get in touch and ask for a free trial.
As ever we hope you find these changes and improvements helpful, we’d love to hear any suggestions you may have, support is available as usual from support@varsome.com
- The VarSome Team
Submit a Comment