
Richard Meyer
CTO
November brings further great improvements to our platform. Many of these improvements have been performed in direct response to feedback from our users and customers.
Note: VarSome will now default to using reference genome hg38 instead of hg19.
Highlights
- CNVs: it is now possible to annotate CNVs directly in VarSome and classify them using the ACMG guidelines.
- ACMG classification:
- Phenotypes: classification now includes the Inclusion of optional patient disease & phenotype information for germline queries.
- Disabling clinical evidence: it is now possible to disable clinical evidence (rules PS3, BS3, PP3 & BP4) to see what the stand-alone classification of the variant would be.
- Multiple refinements to the ACMG classifier resulting in fewer false positives.
- AMP: The classifier now identifies approved therapies that match a patient's cancer type, and provides a table listing the details of those therapies. All information is retrieved from the Jax CKB database.
- Publication tags: now include drug names, diseases and phenotypes.
- A vast number of fixes & improvements to the VarSome & VarSome Clinical platforms.
As always, we hope these changes reflect our company mission statement “To enable anyone to find, share, use the most comprehensive human genome data - and to collaborate to improve health and lives around the world.” and we continue to value your feedback so that we can continue to improve in all areas of development.
ACMG CNV Classification in VarSome
We are very excited to announce that it is now possible to classify CNVs in VarSome:
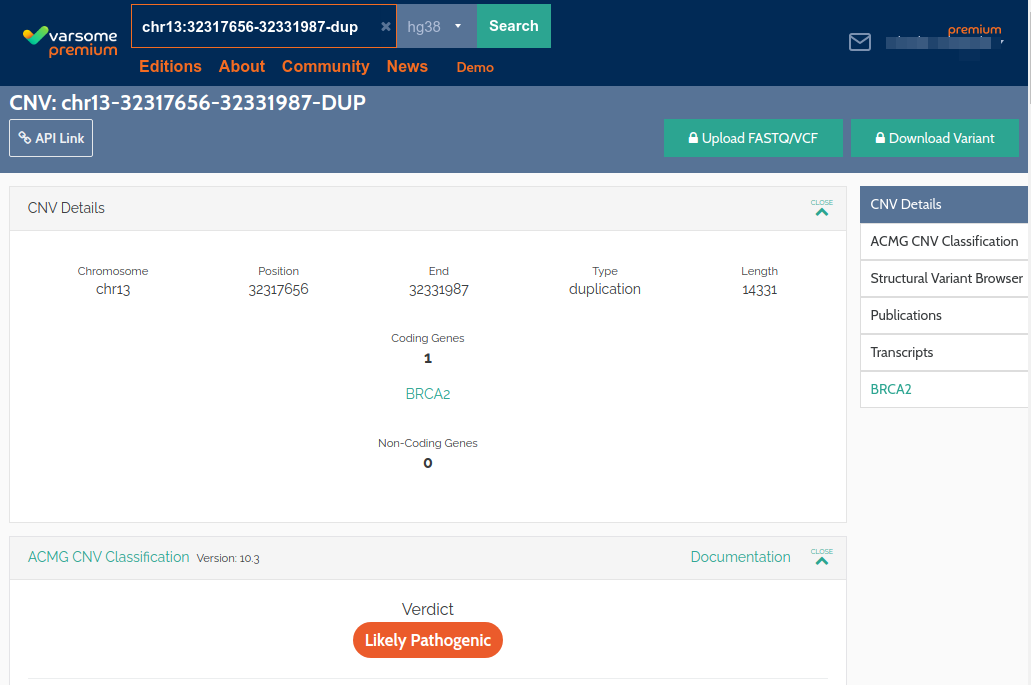
Please see here for more complete information.
ACMG Classifier
Phenotypes
To improve your ACMG classification for germline variants you can now enter related patient phenotype information, in the same way that you can currently do for somatic variants:
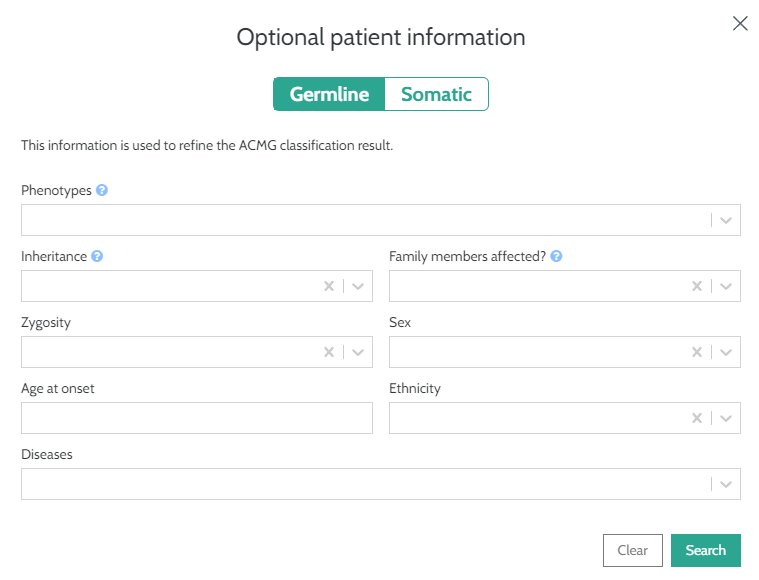
Please see here for more complete information.
Disabling Clinical Evidence
In addition to the above, it is now possible to disable clinical evidence in the ACMG results in the front-ends of the VarSome & VarSome Clinical platforms. In this case the clinical evidence rules (PS3, BS3, PP5 & BP6) will still be evaluated, but will not be taken into account by the verdict. This allows you to see whether the classification would still be supported without any external clinical evidence.
The following screenshot shows the ACMG verdict with clinical evidence disabled, notice how rule PP5 has been deactivated, however it still has a red highlight and the evidence is still visible. In this example the verdict changes from Pathogenic to Likely Pathogenic as a result:
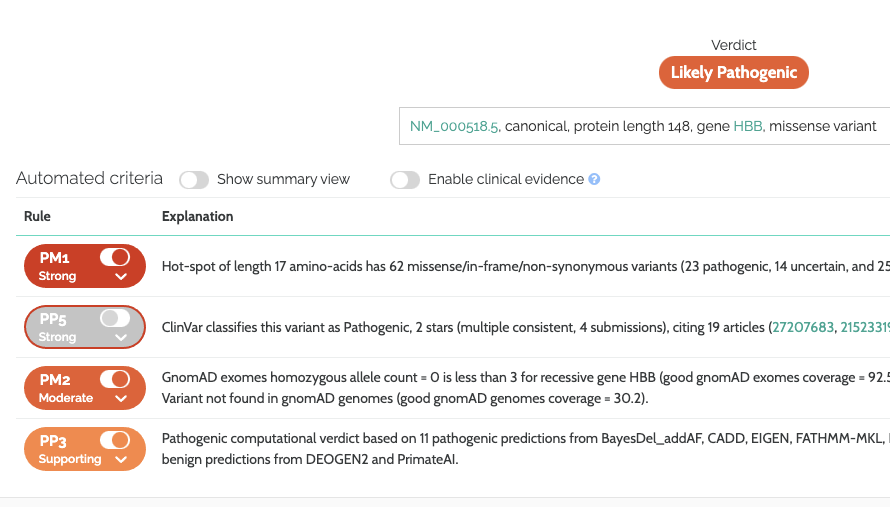
Improved Calibration
Based on user feedback, we have refined our ACMG classifier in order to significantly reduce the number of false positives (variants incorrectly classified as Pathogenic or Likely Pathogenic) benign, at the expense of a minimal number of false negatives (variants incorrectly classified as Benign or Likely Benign):
- Rule PM2 now takes conservation into account in order to reduce the number of false positives (variants incorrectly classified as pathogenic). The rule now triggers with strength “supporting” instead of the default “moderate” if the position is not conserved according to PhyloP. It will also trigger with strength “strong” if the position is highly conserved.
- Rule PM1: is now less aggressive, resulting in fewer false positives. It triggers with strengths “supporting”, “moderate” and “strong” depending on the density of the hot-spot or pathogenicity of the protein domain considered.
- Rules PP3 & BP4: will no longer trigger in edge cases where we have approximately the same number of benign and pathogenic in-silico predictions.
- Rule BP7: uses a slightly higher threshold for PhyloP conservation, resulting in more VUS or Likely Benign verdicts.
- Rule PVS1: we reduce the strength to strong for a slightly higher percentage of last exon deleted.
- Rules PP2 & BP1: the gene missense thresholds used have been adjusted, resulting in more VUS variants.
These changes have been calibrated & tested against over a million reliable classifications - they are the result of statistical analysis of this large dataset.
VarSome Improvements
New Databases
We have now incorporated FusionGDB into VarSome. This database provides functional annotation of fusion genes in cancer, providing better therapeutic targets:
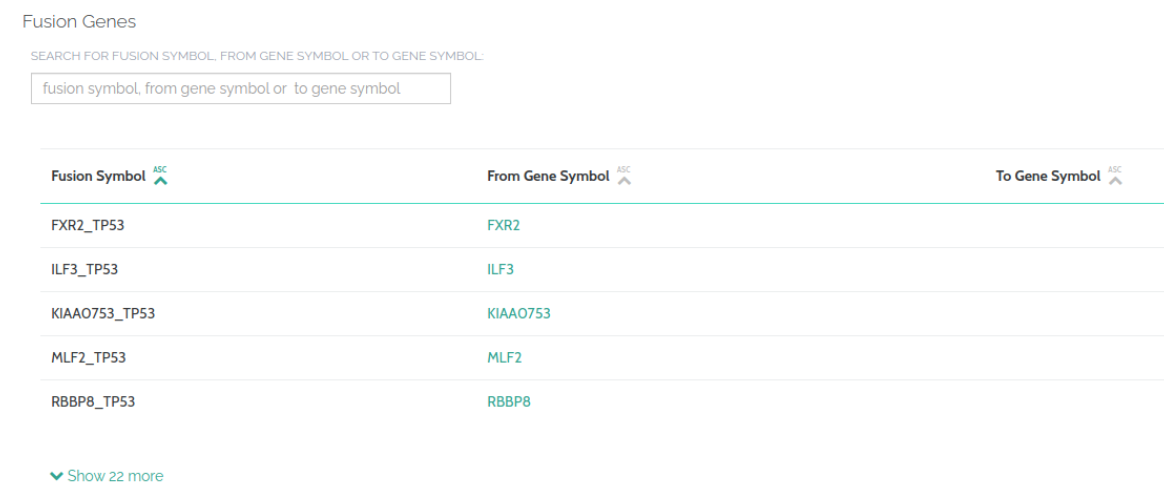
The full list of Data Sources can be found here.
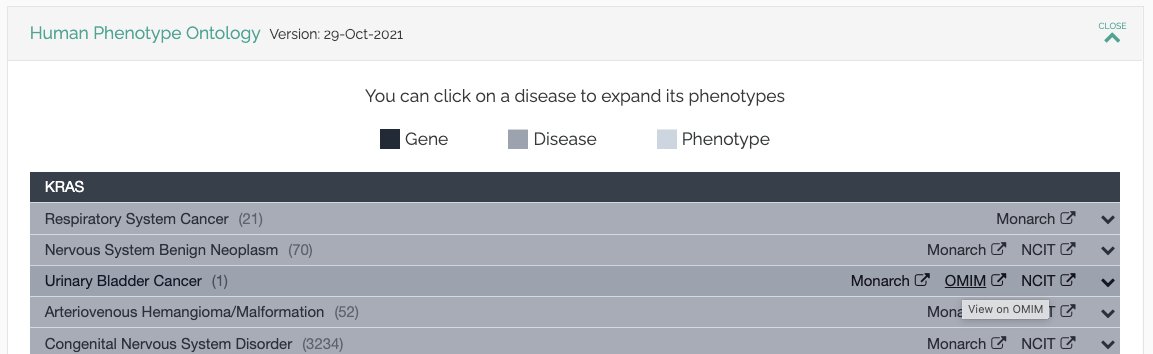
OMIM
We have enhanced our integration with OMIM, leveraging the identifiers provided by HPO and MONDO to link to the OMIM website:
Default to hg38 instead of hg19
The default reference genome has now been set to hg38 instead of hg19. This can be overridden on the various search screens:

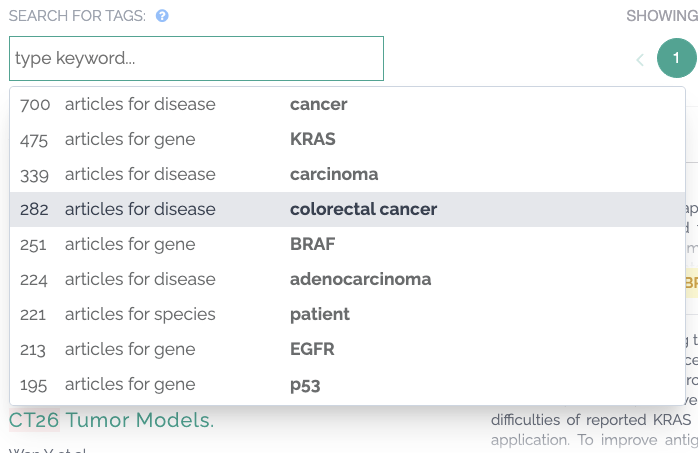
Publications Improvements
We have introduced tagging for drugs and phenotypes using curated sources such as EMA, FDA and HPO.
We have also improved disease tagging by taking into consideration Mondo Disease Ontology disease definitions.
You can use the search functionality to view number of articles by tag:
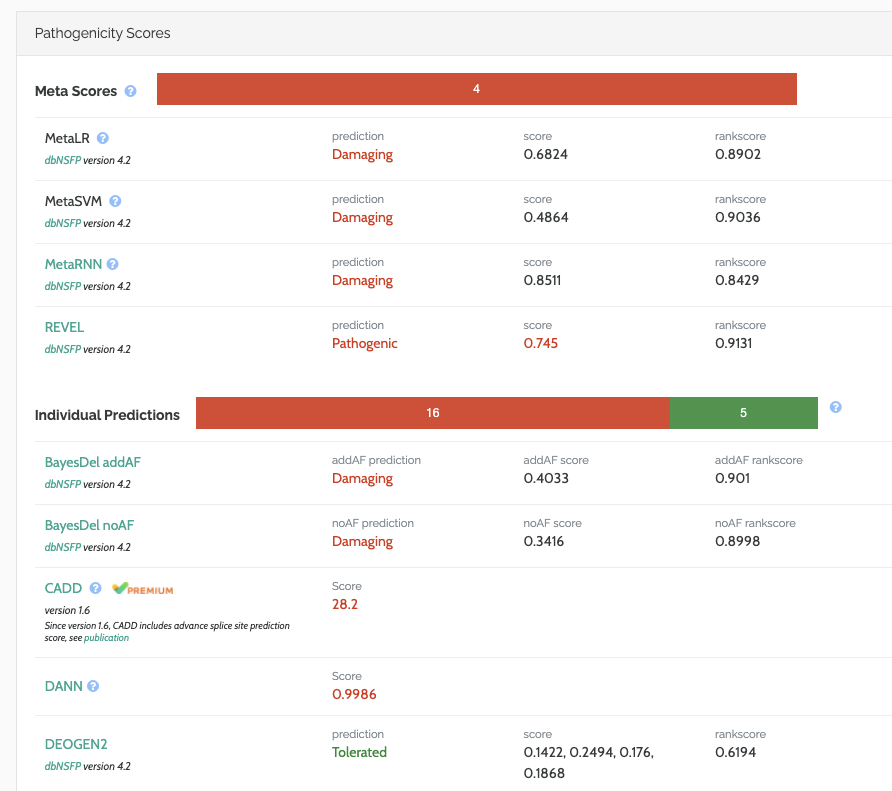
In-Silico Predictions
We’ve improved the display of in-silico predictions, splitting out the ‘meta’ scores which combine the evidence from multiple individual predictors:
VarSome API
We have now included CNVs into our VarSome API. Further information about updates to our API can be seen in the API release note.
The add-all-data query parameter is now deprecated and functions as a shortcut for add-source-databases=all
Queries on the API will now validate request parameters and raise a 400 (Bad request error) in case a parameter has not the expected value. E.g. add-source-databases=dummy-database will raise a 400 Bad request error.
For information regarding the request parameters and their possible values you can read through documentation provided for Variant Queries , Gene Queries , CNV Queries .
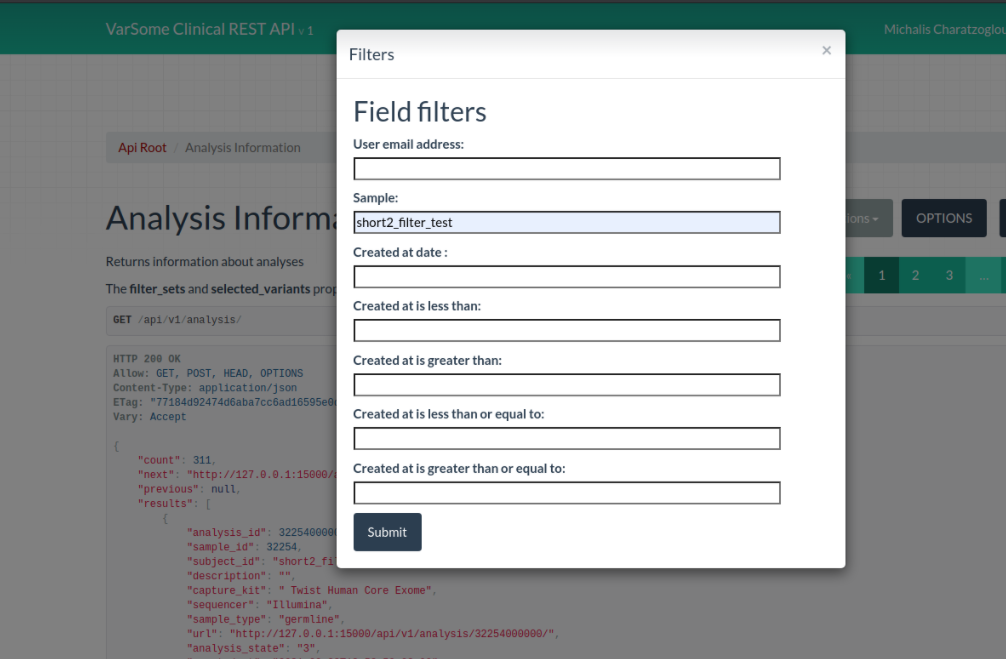
VarSome Clinical API
A new Field Filter has been added to the API so that you can now search by Sample Name.
VarSome Clinical
The following improvements have been made to VarSome Clinical, our platform for FASTQ and VCF analysis.
AMP Classifier - Approved Therapies
In the VarSome clinical platform, when JAX CKB is enabled, the AMP classifier now identifies approved therapies that match a patient's cancer type:
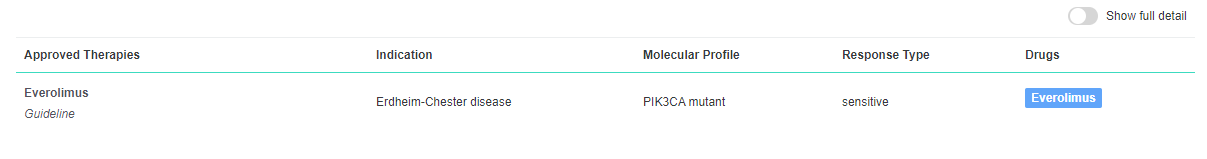
By clicking the “Show full detail” toggle button you can expand the table and additional information is displayed:

Please Note: Importantly these are not recommendations for treatment, we are simply highlighting which entries have been identified in the Jax CKB database.
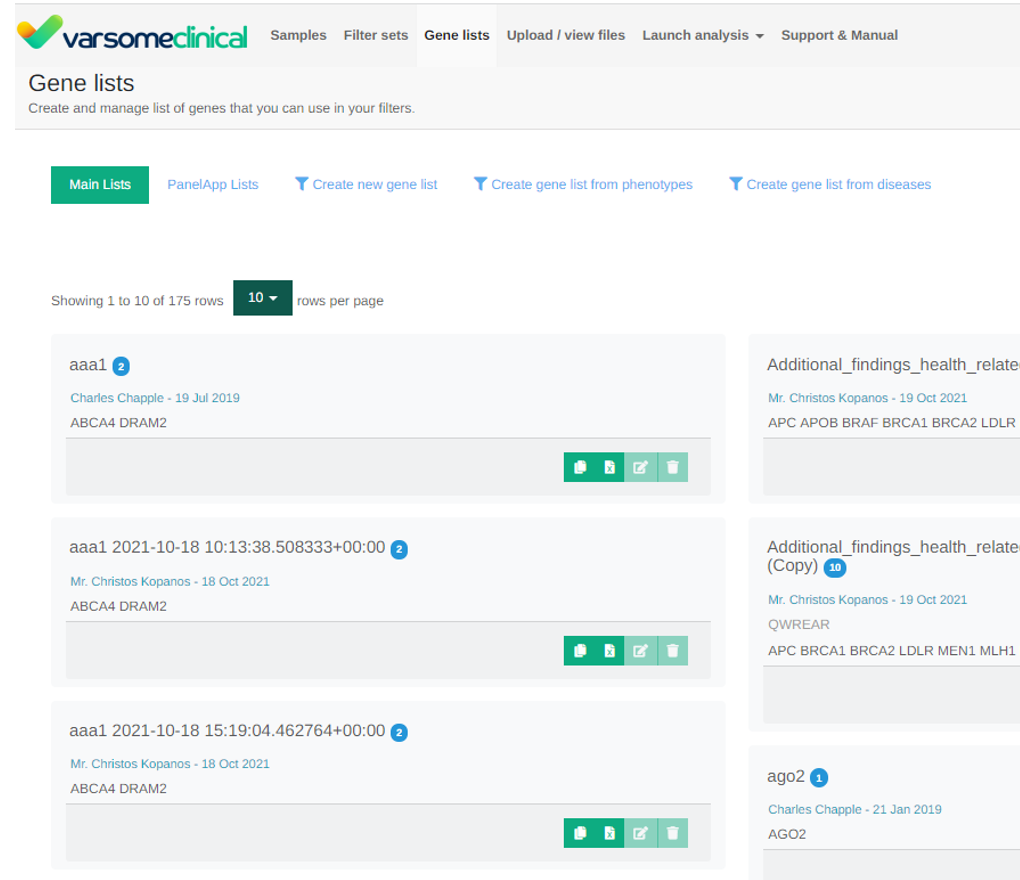
Gene Lists
Improvements to the gene list display screen by introducing pagination and also we have made it faster to create and load gene lists:
Coding Coverage
It is now possible to download a coding coverage report on gene panels for sub-analysis of trios:

Somatic Analysis from FastQ
We have added a new radio button option on the launch somatic analysis from FASTQ that allows you to choose whether to see all variants, or only those that pass the quality filters. This was previously only available for germline analyses:

CNV filtering for mixed-sex cohorts
VarSome Clinical will no longer filter out CNVs on the sex chromosomes in analyses of mixed sexes. This had been done with the previous release to avoid returning unreliable CNV calls, since a cohort of mixed sexes can cause false positives to be called in the sex chromosomes. However, some of our users have told us they prefer to see these calls and decide whether to believe them or not so we are reverting the change.
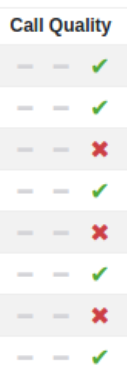
CNV Quality Scores
Call Quality visualisation for CNV has been improved using ticks and “X”s, so at a glance you can see which has passed and failed the quality checks:
Custom Classifications for CNVs
It is now possible to manually classify CNVs in the same way as for small variants:
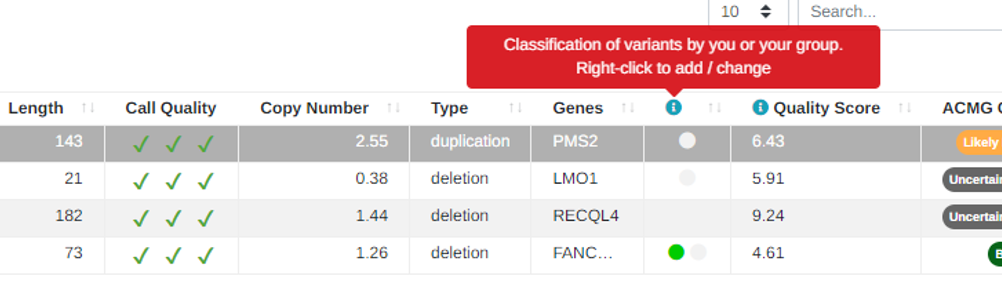
For custom classifications of both small variants and CNVs we also now show the last date the variant was interpreted so it is possible to see when the custom classification was last changed:
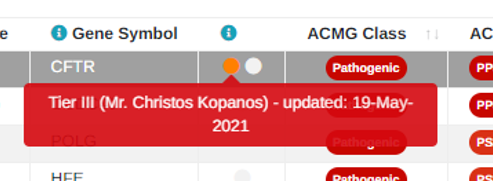
Custom classifications have also been added to the excel spreadsheet along with the name of who the classification was added by.
VCF Filters
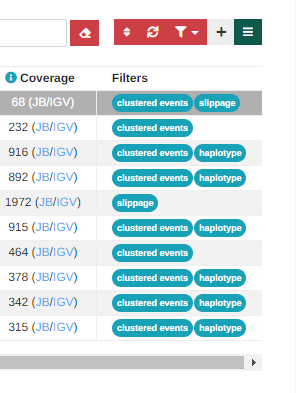
We have added a new “Filters” column on the analysis results page which shows the specific filters that a variant has failed. The information is taken from the source VCF file, so if the analysis has been run from VCF data, any filters present in the input file will be shown in the column.
This column will not show filters for legacy analyses, run before this change was implemented:
Security
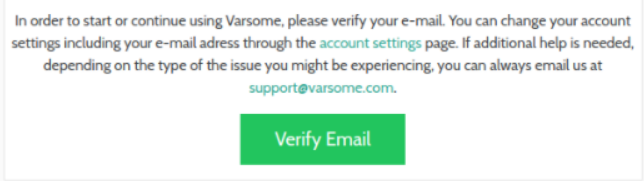
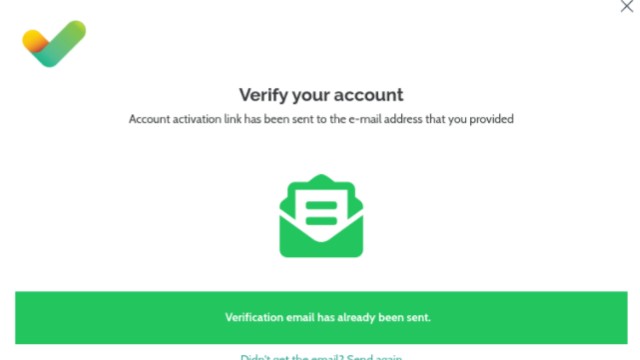
We have improved our security so you now need to verify your Email address in order to start or continue using VarSome:
Further Information and Support
Not already a VarSome Premium, VarSome API or VarSome Clinical user? Get in touch and ask for a free trial.
As ever we hope you find these changes and improvements helpful, we’d love to hear any suggestions you may have, support is available as usual from support@varsome.com
- The VarSome Team
Submit a Comment