
Richard Meyer
CTO
VarSome v10.2 several significant enhancements to our platform:
- CNV Visualization & Quality Scores: the most significant addition to our Clinical platform is the provision of a new visualisation tool for CNVs; we also now flag CNV calls that may be problematic (e.g. low coverage or difficult to map genomic region).
- Submissions to ClinVar: you can now submit variants to ClinVar directly from the VarSome platform (you need to register with ClinVar first) using their new API. You can then keep track of your submissions, approvals, corrections & resubmissions in VarSome.
- New Databases: further significant enhancements to the Varsome free platform are the introduction of three new components GnomAD mitochondrial, Clingen genes and Protein Atlas. We have also improved the display of PharmGKB data.
- ACMG Classifier improvements: we have refined several rules to increase accuracy, and the reporting of clinical evidence now includes the automatic lift-over of user-linked publications, and identifies more functional studies using VarSome AI.
- Our AMP Classifier is now better able at resolving cancer types and identifying the related FDA-approved drugs.
- Publications are now sorted by Relevance: taking into the account the number of citations, and the variants and genes mentioned.
- User Messaging: building on the success of the VarSome Community contributions, we have added a new Messaging system. This provides the ability to immediately message another VarSome community member to share information, views and ideas. Note: Messaging is currently running quite slowly while the implementation takes place, but should be up to speed soon.
As always, we hope these changes reflect our company mission statement “To enable anyone to find, share, use the most comprehensive human genome data - and to collaborate to improve health and lives around the world.”.
Automated Classifications
ACMG Classifier
There are a number of improvements to the ACMG classifier in this release:
- Automatic lift-over of user-linked publications on hg19 or hg38 to the other genome if equivalent entries don’t already exist. This brings our hg38 classification in-line with hg19.
- Rules PVS1 & BP7 have been enhanced:
- PVS1 will always trigger if +/-2 bases from the canonical splice point. Furthermore the strength is reduced to Strong if the variant is in the last exon and removes less than 1.5% of the protein.
- BP7 will now trigger for synonymous variants that are more than 2 bases from the canonical splice-site (was 11 previously).
- Improved identification of Functional Studies: the VarSome AI tagger is now used to identify functional studies mentioned in publications, and has been extended over all sources of clinical evidence (previously only ClinVar) so now works for UniProt & MitoMap too. This feature still requires manual curation by the clinician.
- Rules PS1 & PM5: fixed a rare problem where these could incorrectly trigger for multi-allelic equivalent or alternative variants. This very rare case typically occurred over canonical splice-sites, the variants would trigger PVS1 anyway so the clinical impact is insignificant.
AMP Classifier
In order to ensure that the patient’s cancer type is correctly identified, the AMP classifer now uses the OncoTree ontology for all clinical data reported from any cancer or gene databases.
This is a big qualitative change to improve the AMP Drug rule in particular:
- Tier I is triggered if there is a drug associated with the patient’s cancer type (or no cancer type is provided).
- Tier II is triggered for drugs associated to the gene, but not specifically related to the patient’s cancer type.
We have also reviewed the AACT clinical trials database to ensure trials, cancer & genes are correctly mapped.
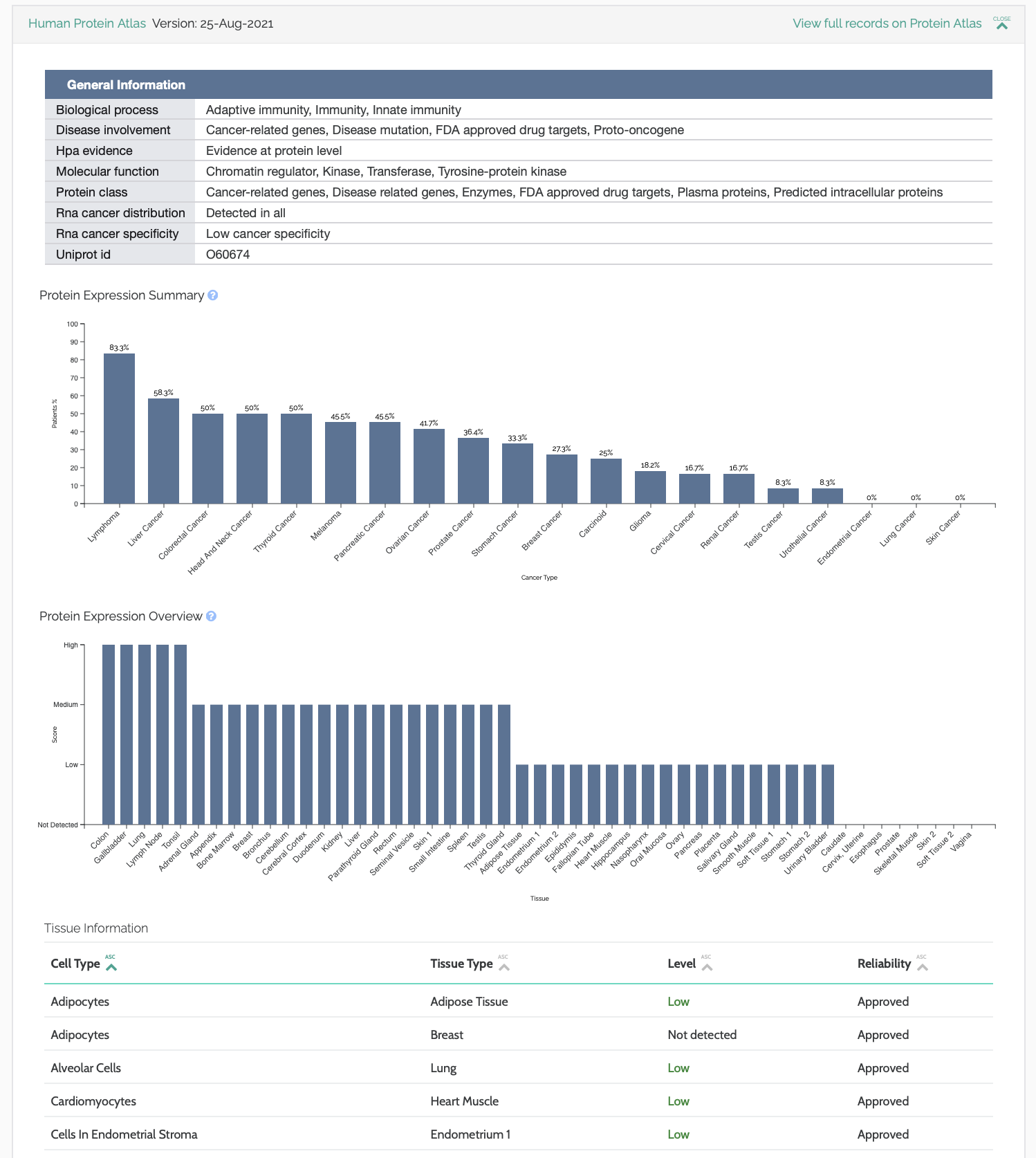
Protein Atlas
The Human Protein Atlas database is now incorporated into VarSome and leveraged by the AMP classifier for the Pathway rule. This Swedish-based open access program aims to map all human proteins in cells, tissues and organs.
The implication for VarSome is that some genes, such as TTN are now being classified as related to cancer, which was not previously the case using the JAX, PMKB & Sanger databases.
VarSome displays general information about the protein, as well as its expression in tissues, and frequency in various cancers:
CNV Quality Flags
We have added a new 'traffic light' system for flagging CNV calls which may be problematic. We will flag (red traffic light) calls if the coverage of the test sample is too low, if the CNV overlaps with one of the 'camouflaged' regions of the genome (see Ebbert et al. 2019) or if the number of selected control samples is low. For further information please see the CNV Quality Help Guide.
VarSome API
We have now included CNVs into our VarSome API. Please contact us if you require further information.
VarSome Clinical
Reuse FASTQ files from multi-sample analyses
We have expanded the option to reuse FASTQ files, from multi-sample analyses as well. A blue box message in the upper right corner of the page will let you know that you will receive an email when the input files are again available for new analysis and which you can view in the "Upload / view files" page.
Visualisation of CNVs
An interactive browser for visualizing CNV results from multi-sample FASTQ CNV analyses has been introduced in the analysis results page. It displays a wider region around the position of the CNV call as well as its location on the chromosome level. The user can zoom in and out using the mouse scroll and select among different chromosomes, genomic locations, samples and CNV calls. Data points represent read ratios (observed/expected read counts) and CNV calls are highlighted. There is an additional coverage track, indicating the trend of the coverage across all cohort samples, as well as genomic annotations (exons/introns). On the right of the graph there is useful CNV call information including genomic location and span, as well as links to the same region in other analyses of the same cohort. For further information please see here or refer to the User Manual.
Structural Variant Browser
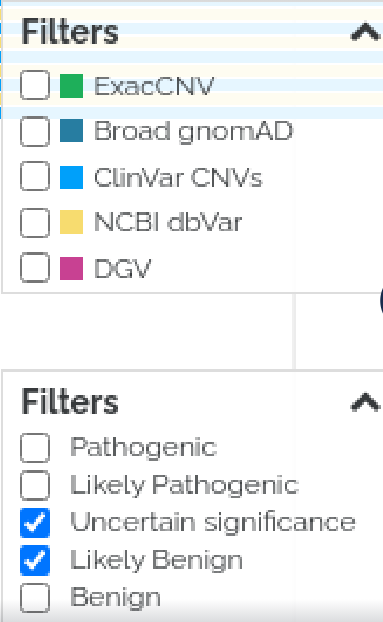
Further improvements have been made to our structural variant browser for CNVs. We have
provided the ability to:
- Filter by Database option
- Filter by Pathogenicity: Pathogenic, Likely Pathogenic, Uncertain, Likely Benign, Benign - allow multiple choices simultaneously (ie: P + LP, or B + LB).
- Blue question marks on aspects that are unclear such as “mean ratio” to provided additional help
VarSome Improvements
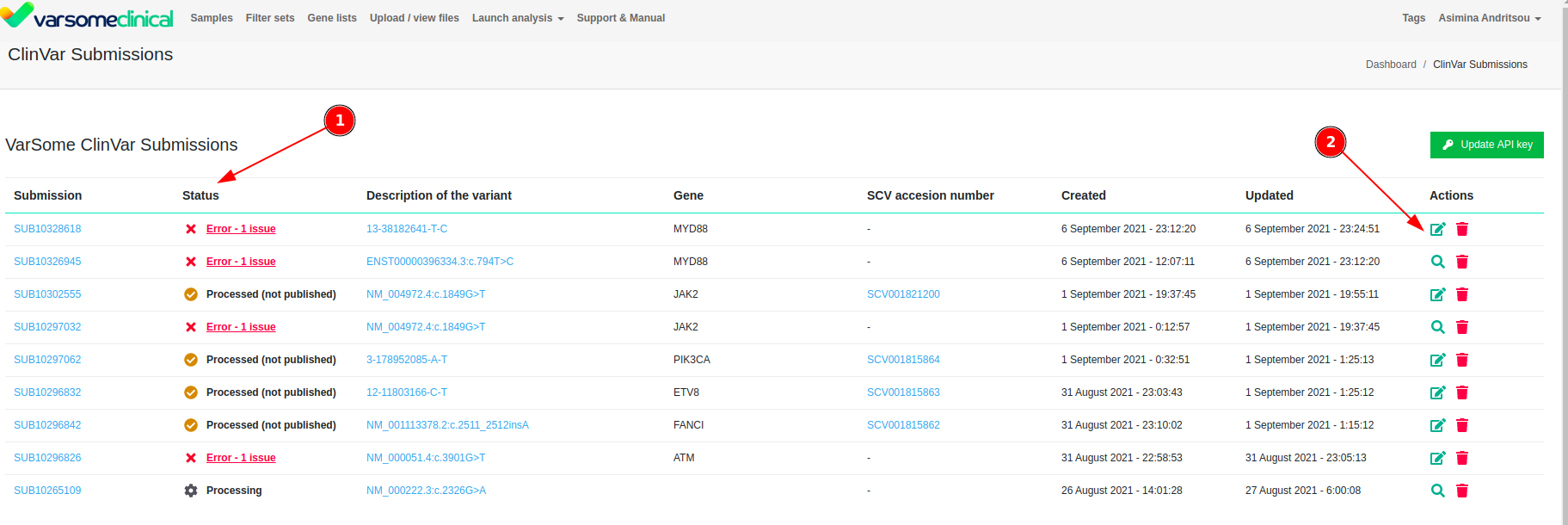
Submissions To ClinVar
It is now possible to submit variants for inclusion in ClinVar directly from the VarSome or VarSomeClinical.
We are excited to announce that we are furthering the collaboration with ClinVar by enabling our users to create ClinVar submissions directly from the VarSome and VarSome Clinical interface.
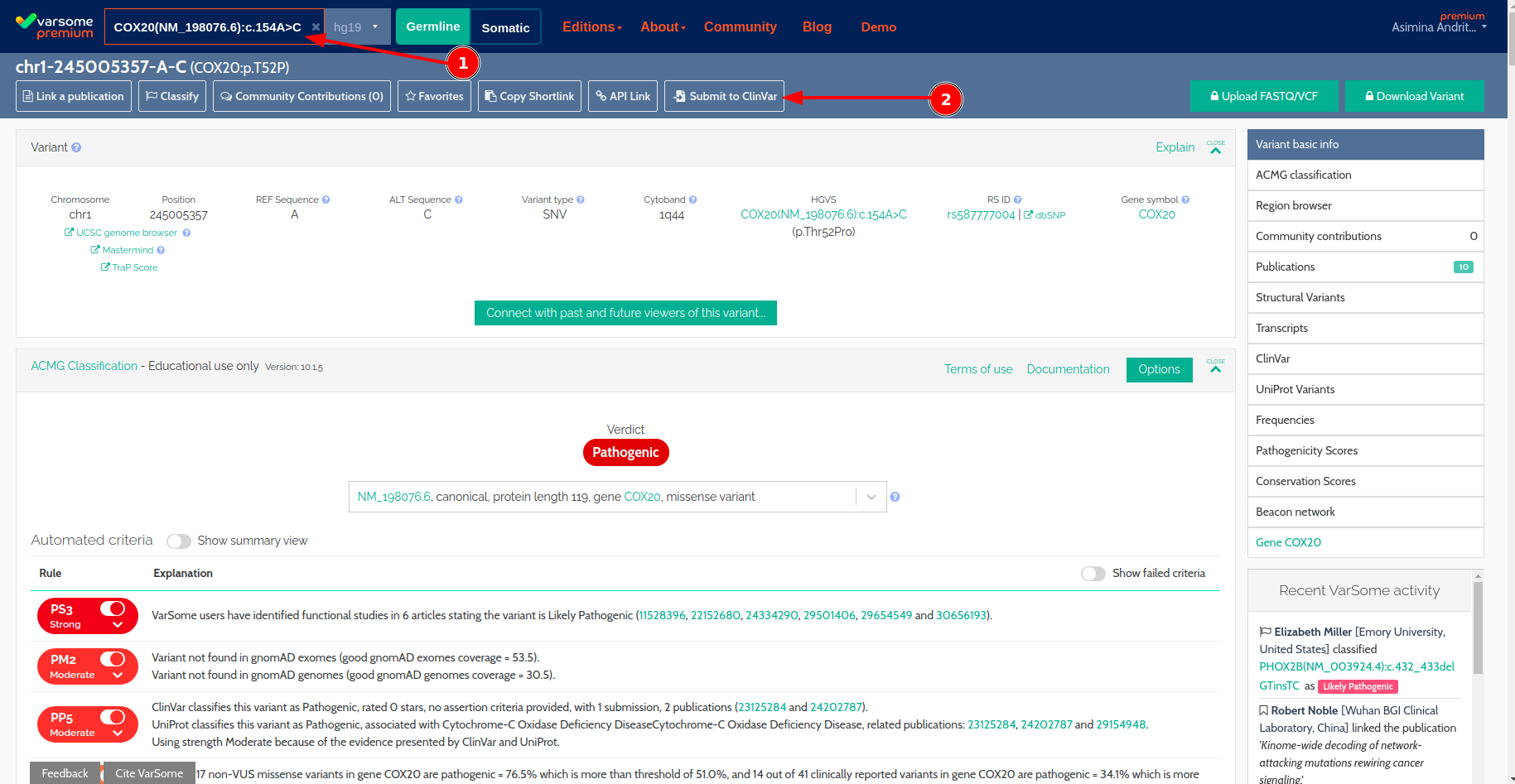
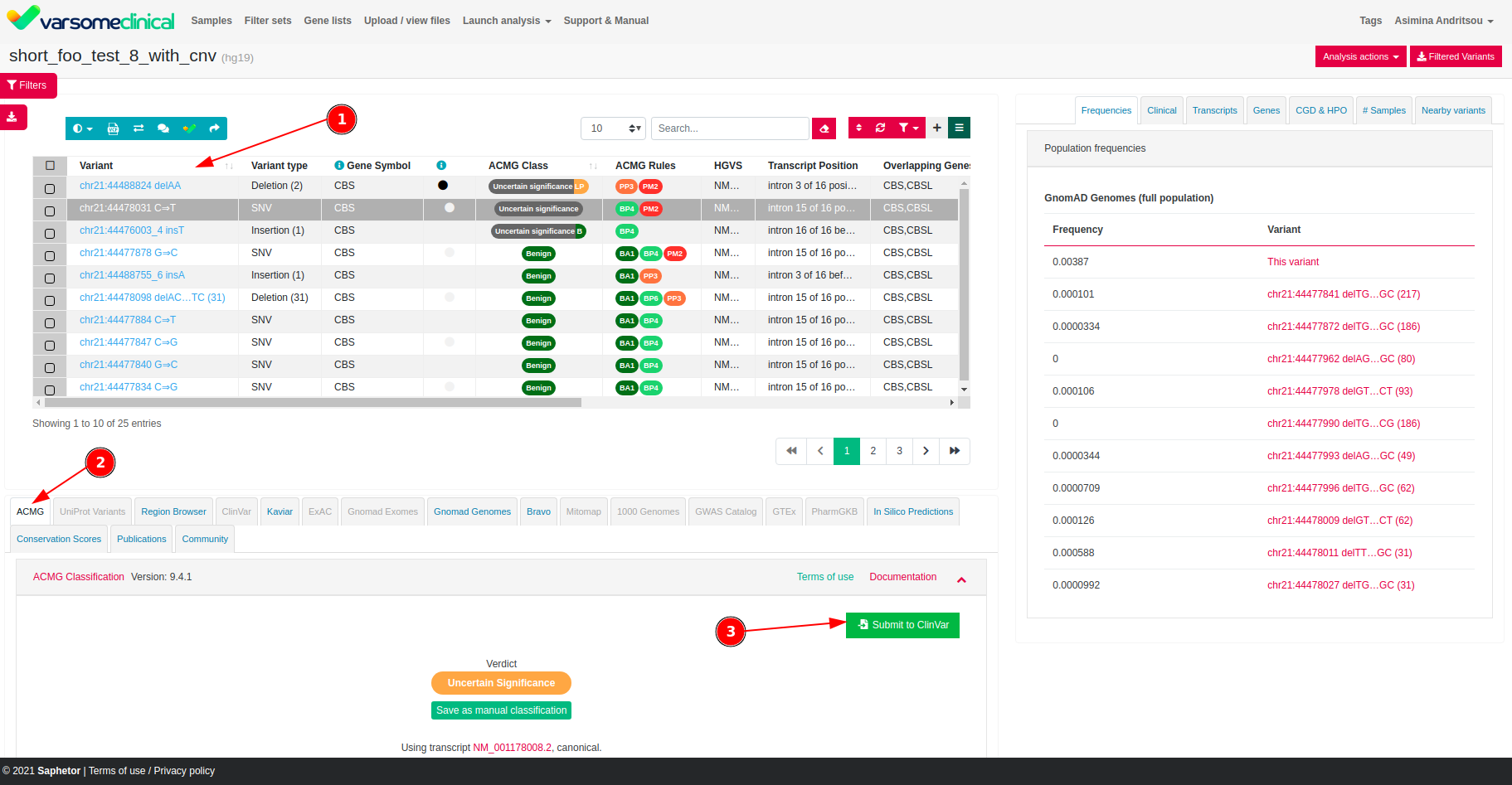
This new feature will allow you not only to submit information for the variant of interest, but also keep track of the list of submissions made by your organization with their processing status and date of creation.
We encourage you to be a part of the extended connection of our users across platforms as well as providing your scientific contribution! More information on how to do this can be found here.
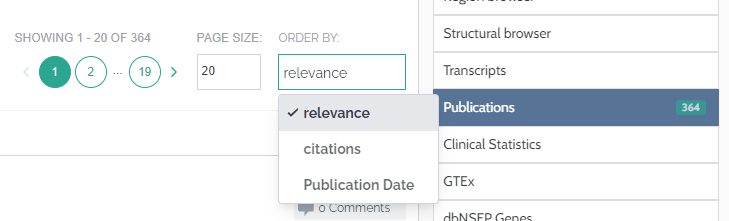
Publications
As an enhancement to our publications component, we now display the amount of times an article has been cited and provide the option to order articles by relevance score or by number of citations. The relevance score considers the particular variant you are looking at, the genes mentioned and the number of citations.
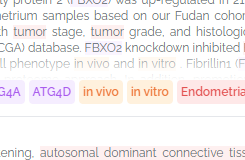
We also now highlight tags for functional studies:
Messaging
Previously it has been possible to view messages sent to you directly from VarSome by clicking on the envelope next to your user name and profile details. In this release we have provided the ability to message any other VarSome community member. You can find further information on this new feature here.
We hope you find this a useful new feature. Currently this is a simple version that we are looking to enhance. We will be grateful for any feedback you may have. To provide feedback please complete this form.
As part of the introduction of messaging we have also improved the User Profile Page to show full organisation data and the ability to upload a photo to personalise your account.
New Databases
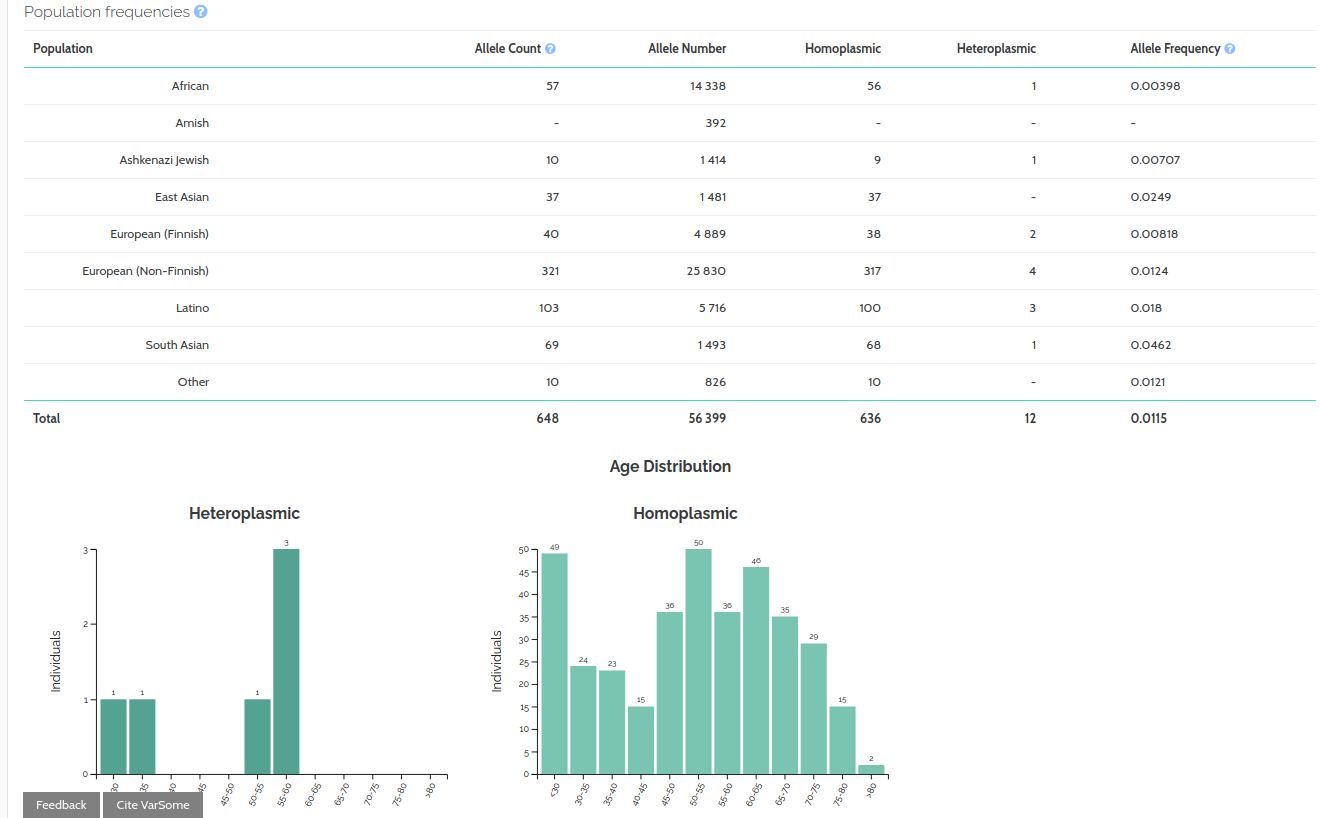
We have also added a new gnomAD Mitochondrial component. Additionally we have added the GnomAD Mitochondrial source to the frequency section of the genome browser.
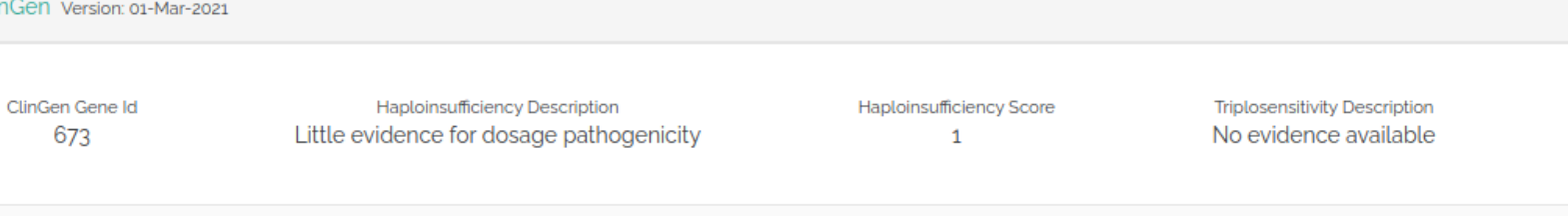
We have created a new component for ClinGen integration (genes)

We have created a newgnomAD Mitochondrial component and additionally have added the GnomAD Mitochondrial source to the region browser frequency section

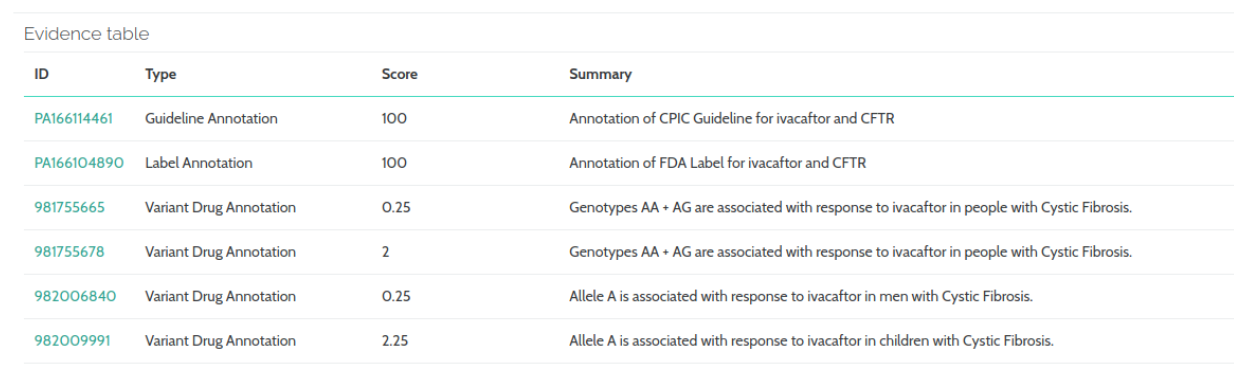
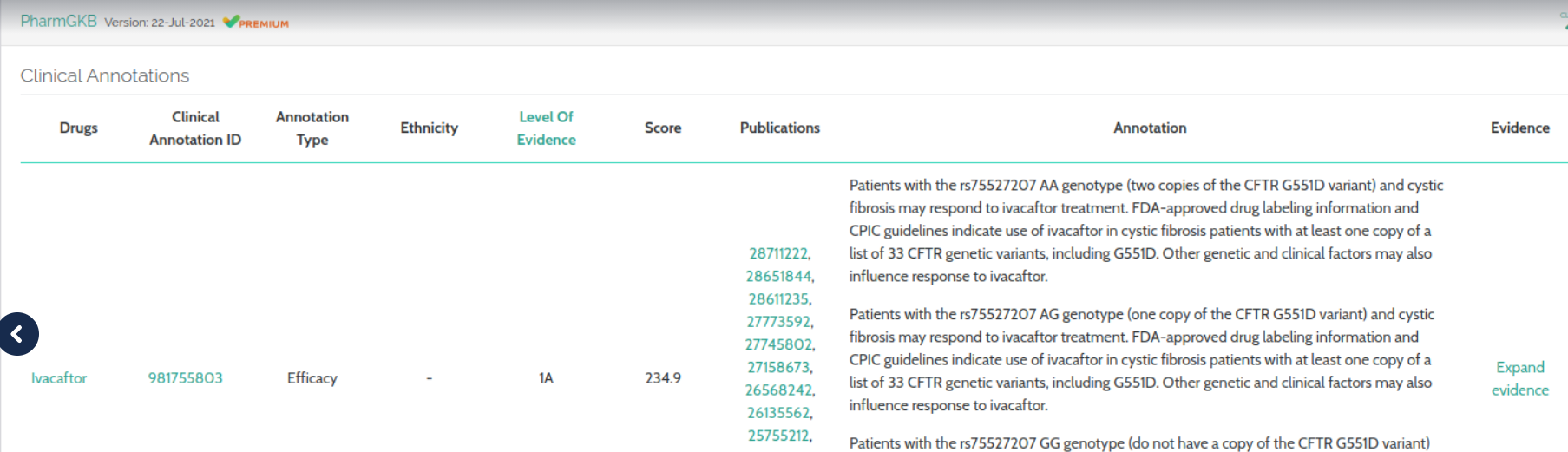
In our PharmGKB Clinical Annotations component, the “ethnicity“ column as been removed, while “url“, “score” and “evidence“ fields have been added.
The full list of Data Sources can be found here.
Further Information and Support
Not already a VarSome Premium or VarSome Clinical user? Get in touch and ask for a free trial.
As ever we hope you find these changes and improvements helpful, we’d love to hear any suggestions you may have, support is available as usual from support@varsome.com
- The VarSome Team
Submit a Comment